Vai trò đa chiều của các cơ quan Ketone
Cơ thể xeton được tạo ra bởi gan và được sử dụng như một nguồn năng lượng khi glucose không có sẵn trong cơ thể con người. Hai thể xeton chính là acetoacetate (AcAc) và 3-beta-hydroxybutyrate (3HB), trong khi axeton là thể xeton thứ ba và ít phong phú nhất. Xeton luôn có trong máu và nồng độ của chúng tăng lên khi nhịn ăn và tập thể dục kéo dài.Ketogenesis là quá trình sinh hóa mà sinh vật tạo ra các cơ quan xeton thông qua sự phân hủy của các axit béo và các axit amin ketogenic.
Ketone cơ thể chủ yếu được tạo ra trong ty thể của tế bào gan. Ketogenesis xảy ra khi có mức đường huyết thấp trong máu, đặc biệt là sau khi các cửa hàng carbohydrate di động khác, chẳng hạn như glycogen, đã cạn kiệt. Cơ chế này cũng có thể xảy ra khi không có đủ lượng insulin. Việc sản xuất các cơ quan xeton được bắt đầu để tạo ra năng lượng sẵn có được lưu trữ trong cơ thể con người như các axit béo. Ketogenesis xảy ra trong ti thể, nơi nó được điều hòa độc lập.
Tóm tắt
Ketone chuyển hóa cơ thể là một nút trung tâm trong homeostasis sinh lý. Trong tổng quan này, chúng tôi thảo luận về cách xeton phục vụ các vai trò trao đổi chất tinh chỉnh rời rạc nhằm tối ưu hóa hoạt động của cơ quan và sinh vật trong các chất dinh dưỡng khác nhau và bảo vệ khỏi viêm và tổn thương trong nhiều hệ thống cơ quan. Theo truyền thống được xem như chất nền trao đổi chất chỉ có trong giới hạn carbohydrate, các quan sát gần đây nhấn mạnh tầm quan trọng của các cơ quan ketone như các chất trung gian trao đổi chất và tín hiệu quan trọng khi carbohydrate dồi dào. Bổ sung một tiết mục lựa chọn điều trị cho các bệnh về hệ thần kinh, vai trò tiềm năng cho cơ thể ketone trong ung thư đã phát sinh, như có vai trò bảo vệ hấp dẫn trong tim và gan, mở ra các lựa chọn điều trị trong bệnh béo phì và tim mạch. Các tranh cãi về sự trao đổi chất và tín hiệu ketone được thảo luận để điều hòa giáo điều cổ điển với các quan sát hiện đại.
Giới thiệu
Các thể xeton là nguồn nhiên liệu trao đổi chất thay thế quan trọng cho tất cả các lĩnh vực của sự sống, nhân chuẩn, vi khuẩn và vi khuẩn cổ (Aneja et al., 2002; Cahill GF Jr, 2006; Krishnakumar et al., 2008). Chuyển hóa cơ thể xeton ở người đã được tận dụng để cung cấp năng lượng cho não trong những giai đoạn thiếu hụt chất dinh dưỡng theo từng đợt. Các cơ quan xeton được đan xen với các con đường trao đổi chất quan trọng của động vật có vú như? Oxy hóa (FAO), chu trình axit tricarboxylic (TCA), gluconeogenesis, de novo lipogenesis (DNL) và sinh tổng hợp sterol. Ở động vật có vú, thể xeton được sản xuất chủ yếu ở gan từ acetyl-CoA có nguồn gốc từ FAO, và chúng được vận chuyển đến các mô ngoài gan để oxy hóa giai đoạn cuối. Sinh lý học này cung cấp một loại nhiên liệu thay thế được tăng cường bởi thời gian nhịn ăn tương đối ngắn, làm tăng lượng axit béo và giảm lượng carbohydrate sẵn có (Cahill GF Jr, 2006; McGarry và Foster, 1980; Robinson và Williamson, 1980). Quá trình oxy hóa xeton trong cơ thể trở thành một yếu tố góp phần đáng kể vào quá trình chuyển hóa năng lượng tổng thể của động vật có vú trong các mô ngoài gan ở vô số trạng thái sinh lý, bao gồm nhịn ăn, nhịn đói, thời kỳ sơ sinh, sau khi tập thể dục, mang thai và tuân thủ chế độ ăn ít carbohydrate. Tổng nồng độ xeton trong cơ thể lưu hành ở người trưởng thành khỏe mạnh thường biểu hiện dao động sinh học giữa khoảng 100 250 M, tăng lên ~ 1 mM sau khi tập thể dục kéo dài hoặc nhịn ăn 24 giờ, và có thể tích lũy đến 20 mM trong các trạng thái bệnh lý như nhiễm toan ceton do tiểu đường ( Cahill GF Jr, 2006; Johnson và cộng sự, 1969b; Koeslag và cộng sự, 1980; Robinson và Williamson, 1980; Wildenhoff và cộng sự, 1974). Gan của con người sản xuất tới 300 g cơ thể xeton mỗi ngày (Balasse và Fery, 1989), đóng góp từ 5 20% tổng năng lượng tiêu thụ ở trạng thái được cho ăn, nhịn ăn và bỏ đói (Balasse et al., 1978; Cox et al., 2016).
Các nghiên cứu gần đây hiện nay làm nổi bật vai trò bắt buộc đối với các cơ quan ketone trong chuyển hóa tế bào động vật có vú, cân bằng nội môi và báo hiệu dưới nhiều trạng thái sinh lý và bệnh lý khác nhau. Ngoài việc dùng làm nhiên liệu năng lượng cho các mô ngoài da như não, tim, hoặc cơ xương, cơ thể ketone đóng vai trò quan trọng như trung gian báo hiệu, trình điều khiển biến đổi protein (PTM) và điều biến viêm và stress oxy hóa. Trong tổng quan này, chúng tôi cung cấp cả hai quan điểm cổ điển và hiện đại về vai trò pleiotropic của các cơ quan ketone và sự trao đổi chất của chúng.
Tổng quan về trao đổi chất cơ thể Ketone
Tốc độ tạo ceton ở gan được điều chỉnh bởi một loạt các biến đổi sinh lý và sinh hóa của chất béo. Các chất điều hòa chính bao gồm sự phân giải lipid của các axit béo từ triacylglycerol, vận chuyển đến và qua màng sinh chất tế bào gan, vận chuyển vào ty thể thông qua carnitine palmitoyltransferase 1 (CPT1), vòng xoắn?-Oxy hóa, hoạt động chu trình TCA và nồng độ trung gian, tiềm năng oxy hóa khử và các chất điều hòa nội tiết tố trong số các quá trình này, chủ yếu là glucagon và insulin [được xem xét trong (Arias và cộng sự, 1995; Ayte và cộng sự, 1993; Ehara và cộng sự, 2015; Ferre và cộng sự, 1983; Kahn và cộng sự, 2005; McGarry và Foster , 1980; Williamson và cộng sự, 1969)]. Sự tạo xeton cổ điển được xem như một con đường lan tỏa, trong đó acetyl-CoA có nguồn gốc từ oxy hóa vượt quá hoạt tính tổng hợp citrate và / hoặc sự sẵn có của oxaloacetate để ngưng tụ tạo thành citrate. Các chất trung gian ba carbon thể hiện hoạt tính chống ketogenic, có lẽ là do khả năng mở rộng vùng oxaloacetate để tiêu thụ acetyl-CoA, nhưng nồng độ acetyl-CoA ở gan không quyết định tỷ lệ ketogenic (Foster, 1967; Rawat và Menahan, 1975; Williamson và cộng sự, 1969). Cơ chế điều hòa tạo xeton bằng các sự kiện nội tiết tố, phiên mã và sau dịch mã hỗ trợ quan điểm rằng các cơ chế phân tử điều chỉnh tốc độ sinh xeton vẫn chưa được hiểu đầy đủ (xem Quy định về HMGCS2 và SCOT / OXCT1).
Quá trình tạo xeton chủ yếu xảy ra trong chất nền ty thể gan với tốc độ tỷ lệ thuận với tổng quá trình oxy hóa chất béo. Sau khi vận chuyển chuỗi acyl qua màng ty thể và?-Oxy hóa, đồng phân ty thể của 3-hydroxymethylglutaryl-CoA synthase (HMGCS2) xúc tác cho số phận gây ra sự ngưng tụ của acetoacetyl-CoA (AcAc-CoA) và acetyl-CoA để tạo ra HMG-CoA (Hình. 1A). HMG-CoA lyase (HMGCL) phân cắt HMG-CoA để giải phóng acetyl-CoA và acetoacetate (AcAc), và sau đó được khử thành d -? - hydroxybutyrate (d-? OHB) bởi ty thể phụ thuộc phosphatidylcholine d-? OHB dehydrogenase ( BDH1) trong phản ứng gần cân bằng kết hợp NAD + / NADH (Bock và Fleischer, 1975; LEHNINGER và cộng sự, 1960). Hằng số cân bằng BDH1 hỗ trợ sản xuất d-? OHB, nhưng tỷ lệ các thể xeton AcAc / d-? OHB tỷ lệ thuận với tỷ lệ NAD + / NADH của ty thể, và do đó hoạt động của BDH1 oxidoreductase điều chỉnh tiềm năng oxy hóa khử của ty thể (Krebs và cộng sự, 1969; Williamson và cộng sự, 1967). AcAc cũng có thể tự động khử cacboxylat thành axeton (Pedersen, 1929), nguồn gốc của mùi ngọt ở người bị nhiễm toan ceton (tức là tổng số thể xeton trong huyết thanh> ~ 7 mM; AcAc pKa 3.6,? OHB pKa 4.7). Cơ chế mà các thể xeton được vận chuyển qua màng trong ti thể vẫn chưa được biết, nhưng AcAc / d-? OHB được giải phóng từ tế bào thông qua các chất vận chuyển monocarboxylate (ở động vật có vú, MCT 1 và 2, còn được gọi là chất mang chất tan 16A các thành viên họ 1 và 7) và được vận chuyển trong tuần hoàn đến các mô ngoài gan để oxy hóa giai đoạn cuối (Cotter và cộng sự, 2011; Halestrap và Wilson, 2012; Halestrap, 2012; Hugo và cộng sự, 2012). Nồng độ của các thể xeton trong tuần hoàn cao hơn nồng độ trong các mô ngoài gan (Harrison và Long, 1940) cho thấy các thể xeton được vận chuyển xuống một gradient nồng độ. Các đột biến mất chức năng trong MCT1 có liên quan đến các đợt nhiễm toan ceton tự phát, cho thấy vai trò quan trọng trong việc nhập ceton vào cơ thể.

Ngoại trừ khả năng chuyển hướng của các thể xeton thành các thể không oxy hóa (xem Các số chuyển hóa không oxy hóa của các thể xeton), tế bào gan không có khả năng chuyển hóa các thể xeton mà chúng tạo ra. Các cơ quan xeton được tổng hợp de novo bởi gan được (i) dị hóa trong ti thể của các mô ngoài gan thành acetyl-CoA, có sẵn cho chu trình TCA cho quá trình oxy hóa cuối (Hình 1A), (ii) chuyển hướng sang con đường tổng hợp lipogenesis hoặc sterol ( Hình 1B), hoặc (iii) bài tiết qua nước tiểu. Là một loại nhiên liệu năng lượng thay thế, các thể xeton được oxy hóa mạnh mẽ trong tim, cơ xương và não (Balasse và Fery, 1989; Bentourkia và cộng sự, 2009; Owen và cộng sự, 1967; Reichard và cộng sự, 1974; Sultan, 1988 ). BDH1 ty thể ngoài gan xúc tác phản ứng đầu tiên của quá trình oxy hóa? OHB, chuyển nó thành AcAc trở lại (LEHNINGER và cộng sự, 1960; Sandermann và cộng sự, 1986). Một d-? OHB-dehydrogenase (BDH2) trong tế bào chất chỉ có 20% nhận dạng trình tự thành BDH1 có Km cao đối với các thể xeton, và cũng đóng một vai trò trong cân bằng nội môi của sắt (Davuluri và cộng sự, 2016; Guo và cộng sự, 2006) . Trong chất nền ty thể ngoài gan, AcAc được kích hoạt thành AcAc-CoA thông qua trao đổi gốc CoA từ succinyl-CoA trong một phản ứng được xúc tác bởi CoA transferase độc nhất của động vật có vú, succinyl-CoA: 3-oxoacid-CoA transferase (SCOT, CoA transferase; được mã hóa bởi OXCT1), thông qua một phản ứng gần cân bằng. Năng lượng tự do được giải phóng khi thủy phân AcAc-CoA lớn hơn năng lượng của succinyl-CoA, tạo điều kiện cho sự hình thành AcAc. Do đó, dòng oxy hóa cơ thể xeton xảy ra do tác động của khối lượng: nguồn cung cấp dồi dào AcAc và tiêu thụ nhanh chóng acetyl-CoA thông qua tổng hợp citrate tạo điều kiện cho sự hình thành AcAc-CoA (+ succinate) bởi SCOT. Đáng chú ý, trái ngược với glucose (hexokinase) và axit béo (acyl-CoA synthetases), việc kích hoạt các thể xeton (SCOT) thành dạng có thể oxy hóa không cần đến ATP. Phản ứng thiolase AcAc-CoA có thể đảo ngược [được xúc tác bởi bất kỳ trong số bốn thiolase ty thể được mã hóa bởi ACAA2 (mã hóa một enzym được gọi là T1 hoặc CT), ACAT1 (mã hóa T2), HADHA hoặc HADHB] tạo ra hai phân tử acetyl-CoA, đi vào chu trình TCA (Hersh và Jencks, 1967; Stern và cộng sự, 1956; Williamson và cộng sự, 1971). Trong các trạng thái xeton (tức là tổng số xeton huyết thanh> 500 M), các thể xeton trở thành những nhân tố đóng góp đáng kể vào việc tiêu thụ năng lượng và được sử dụng trong các mô một cách nhanh chóng cho đến khi sự hấp thụ hoặc bão hòa của quá trình oxy hóa xảy ra (Balasse và cộng sự, 1978; Balasse và Fery, 1989 ; Edmond và cộng sự, 1987). Một phần rất nhỏ các thể xeton có nguồn gốc từ gan có thể được đo dễ dàng trong nước tiểu, và tỷ lệ sử dụng và tái hấp thu của thận tương ứng với nồng độ tuần hoàn (Goldstein, 1987; Robinson và Williamson, 1980). Trong các trạng thái xeton cao (> 1 mM trong huyết tương), keton niệu đóng vai trò như một báo cáo bán định lượng của ketosis, mặc dù hầu hết các xét nghiệm lâm sàng về thể xeton trong nước tiểu đều phát hiện được AcAc nhưng không phát hiện được OHB (Klocker và cộng sự, 2013).
Ketogenic Substrates và tác động của chúng lên sự trao đổi chất của tế bào gan
Chất nền Ketogenic bao gồm axit béo và axit amin (Hình. 1B). Sự dị hóa của các axit amin, đặc biệt là leucine, tạo ra khoảng 4% các cơ quan xeton ở trạng thái sau hấp thụ (Thomas và cộng sự, 1982). Do đó, chất nền acetyl-CoA tạo ra các cơ quan xeton chủ yếu xuất phát từ các axit béo, bởi vì trong quá trình cung cấp carbohydrate giảm, pyruvate đi vào chu trình TCA gan chủ yếu thông qua chứng mất an toàn, ví dụ, carboxyl hóa phụ thuộc ATP với oxaloacetate (OAA) hoặc (MAL), và không oxy hóa decarboxyl hóa thành acetyl-CoA (Jeoung et al., 2012; Magnusson và cộng sự, 1991; Merritt và cộng sự, 2011). Trong gan, glucose và pyruvate đóng góp không đáng kể vào ketogenesis, ngay cả khi pyruvate decarboxyl hóa thành acetyl-CoA là cực đại (Jeoung et al., 2012).
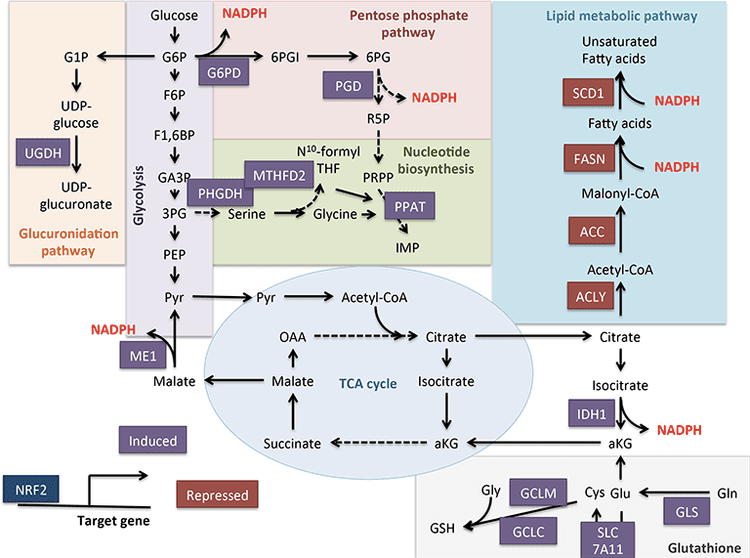
Acetyl-CoA có một số vai trò không thể thiếu trong quá trình trao đổi chất trung gian của gan ngoài thế hệ ATP thông qua quá trình oxy hóa đầu cuối (xem sự tích hợp chuyển hóa cơ thể ketone, thay đổi sau phiên dịch và sinh lý tế bào). Acetyl-CoA allosterically kích hoạt (i) pyruvate carboxylase (PC), do đó kích hoạt một cơ chế kiểm soát trao đổi chất làm tăng sự xâm nhập anaplerotic của các chất chuyển hóa vào chu trình TCA (Owen và cộng sự, 2002; Scrutton và Utter, 1967) và (ii) pyruvate dehydrogenase kinase, mà phosphorylates và ức chế pyruvate dehydrogenase (PDH) (Cooper và cộng sự, 1975), do đó tiếp tục tăng cường dòng chảy của pyruvate vào chu trình TCA thông qua chứng mất an toàn. Hơn nữa, acetyl-CoA tế bào chất, có hồ bơi được tăng cường bởi cơ chế chuyển đổi ty thể acetyl-CoA thành các chất chuyển hóa, ức chế quá trình oxy hóa axit béo: acetyl-CoA carboxylase (ACC) xúc tác chuyển đổi acetyl-CoA thành malonyl-CoA, chất nền lipogenic và chất ức chế allosteric của CPT1 ty thể [xem xét trong (Kahn và cộng sự, 2005; McGarry và Foster, 1980)]. Do đó, hồ bơi acetyl-CoA ti thể vừa điều hòa vừa được điều hòa bởi quá trình lan truyền ketogenesis, phối hợp các khía cạnh quan trọng của quá trình chuyển hóa trung gian gan.
Số lượng trao đổi chất không oxy hóa của các cơ quan Ketone
Số phận chủ yếu của xeton có nguồn gốc từ gan là quá trình oxy hóa phụ thuộc SCOT. Tuy nhiên, AcAc có thể được xuất khẩu từ ty lạp thể và được sử dụng trong các quá trình chuyển hóa thông qua chuyển đổi sang AcAc-CoA bằng phản ứng phụ thuộc ATP được xúc tác bởi acetoacetyl-CoA synthetase tế bào chất (AACS, Hình. 1B). Con đường này hoạt động trong quá trình phát triển não bộ và ở tuyến vú cho con bú (Morris, 2005; Robinson và Williamson, 1978; Ohgami và cộng sự, 2003). AACS cũng được thể hiện rất rõ trong mô mỡ, và các tế bào hủy hoạt hóa (Aguilo và cộng sự, 2010; Yamasaki và cộng sự, 2016). Actopacasmic AcAc-CoA có thể do HMGCS1 bào tương hướng tới sinh tổng hợp sterol, hoặc được phân tách bởi hai loại thiolase tế bào chất thành acetyl-CoA (ACAA1 và ACAT2), carboxyl hóa thành malonyl-CoA, và góp phần tổng hợp các axit béo (Bergstrom et al., 1984; Edmond, 1974; Endemann và cộng sự, 1982; Geelen và cộng sự, 1983; Webber và Edmond, 1977).
Trong khi ý nghĩa sinh lý vẫn chưa được thiết lập, xeton có thể đóng vai trò như chất nền đồng hóa ngay cả trong gan. Trong bối cảnh thí nghiệm nhân tạo, AcAc có thể đóng góp tới một nửa lượng lipid mới được tổng hợp và lên đến 75% lượng cholesterol mới được tổng hợp (Endemann và cộng sự, 1982; Geelen và cộng sự, 1983; Freed và cộng sự, 1988). Bởi vì AcAc có nguồn gốc từ quá trình oxy hóa chất béo ở gan không hoàn toàn, khả năng của AcAc góp phần vào quá trình tạo lipogenesis in vivo sẽ ngụ ý chu trình vô hiệu hóa ở gan, nơi xeton có nguồn gốc từ chất béo có thể được sử dụng để sản xuất lipid, một khái niệm có ý nghĩa sinh lý đòi hỏi phải xác nhận thực nghiệm, nhưng có thể vai trò thích nghi hoặc không thích ứng (Solinas và cộng sự, 2015). AcAc cung cấp rất nhiều cholesterogenesis, với AACS Km-AcAc thấp (~ 50 M) tạo điều kiện cho việc kích hoạt AcAc ngay cả trong trạng thái được cho ăn (Bergstrom và cộng sự, 1984). Vai trò năng động của chuyển hóa xeton trong tế bào chất đã được đề xuất trong tế bào thần kinh phôi chuột sơ cấp và trong tế bào mỡ có nguồn gốc 3T3-L1, vì AACS làm suy giảm sự biệt hóa của từng loại tế bào (Hasegawa và cộng sự, 2012a; Hasegawa và cộng sự, 2012b). Knockdown AACS ở chuột in vivo làm giảm cholesterol huyết thanh (Hasegawa et al., 2012c). SREBP-2, một chất điều hòa phiên mã chính của quá trình sinh tổng hợp cholesterol, và thụ thể hoạt hóa chất tăng sinh peroxisome (PPAR) -? là chất kích hoạt phiên mã AACS, và điều chỉnh quá trình phiên mã của nó trong quá trình phát triển neurite và trong gan (Aguilo và cộng sự, 2010; Hasegawa và cộng sự, 2012c). Tổng hợp lại, chuyển hóa cơ thể xeton trong tế bào chất có thể quan trọng trong một số điều kiện chọn lọc hoặc lịch sử bệnh tật tự nhiên, nhưng không đủ để loại bỏ các thể xeton có nguồn gốc từ gan, vì tăng sinh ceton máu lớn xảy ra trong bối cảnh suy giảm có chọn lọc số phận oxy hóa chính do đột biến mất chức năng. cho SCOT (Berry và cộng sự, 2001; Cotter và cộng sự, 2011).
Quy định về HMGCS2 và SCOT / OXCT1
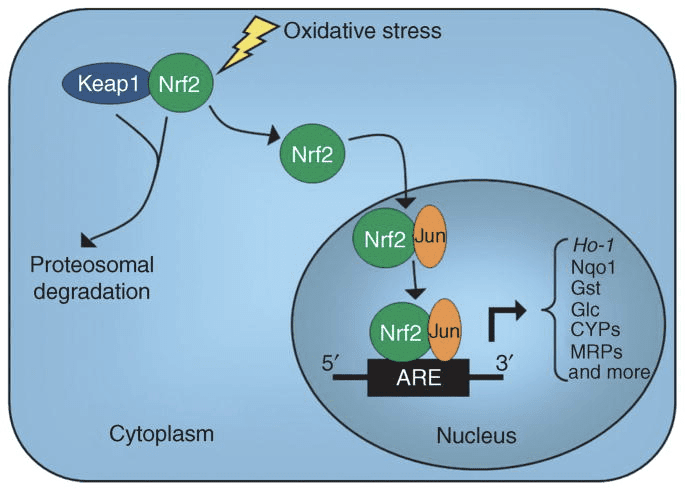
Sự phân kỳ của ty thể từ gen mã hóa cytosolic HMGCS xảy ra sớm trong quá trình tiến hóa động vật có xương sống do nhu cầu hỗ trợ ketogenesis gan ở các loài có tỷ lệ trọng lượng não và cơ thể cao hơn (Boukaftane et al., 1994; Cunnane và Crawford, 2003). Các đột biến HMGCS2 mất chức năng tự nhiên ở người gây ra tình trạng hạ đường huyết hypoketotic (Pitt và cộng sự, 2015; Thompson và cộng sự, 1997). Biểu hiện HMGCS2 mạnh mẽ bị hạn chế đối với tế bào gan và biểu mô ruột, và biểu hiện và hoạt động của enzyme được phối hợp thông qua các cơ chế khác nhau (Mascaro và cộng sự, 1995; McGarry và Foster, 1980; Robinson và Williamson, 1980). Trong khi phạm vi đầy đủ của các trạng thái sinh lý ảnh hưởng đến HMGCS2 đòi hỏi phải làm sáng tỏ thêm, biểu hiện và / hoặc hoạt động của nó được điều chỉnh trong giai đoạn hậu sản sớm, lão hóa, tiểu đường, đói hoặc ăn ketogenic (Balasse và Fery, 1989; Cahill GF Jr, 2006 Girard và cộng sự, 1992; Hegardt, 1999; Satapati và cộng sự, 2012; Sengupta và cộng sự, 2010). Ở bào thai, quá trình methyl hóa vùng 5 bên của gen Hmgcs2 tương quan nghịch với quá trình phiên mã của nó và bị đảo ngược một phần sau khi sinh (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al. ., 1983). Tương tự, Bdh1 gan biểu hiện một mô hình biểu hiện phát triển, tăng từ lúc sinh đến cai sữa, và cũng được gây ra bởi chế độ ăn ketogenic trong một yếu tố tăng trưởng nguyên bào sợi (FGF) -21-phụ thuộc (Badman et al., 2007; Zhang et al., 1989 ). Ketogenesis ở động vật có vú là rất nhạy cảm với cả insulin và glucagon, bị ức chế và kích thích, tương ứng (McGarry và Foster, 1977). Insulin ngăn chặn tình trạng lipipysis mô mỡ, do đó làm mất ketogenesis của chất nền của nó, trong khi glucagon làm tăng thông lượng ketogenic thông qua một hiệu ứng trực tiếp trên gan (Hegardt, 1999). Sự phiên mã Hmgcs2 được kích thích bởi yếu tố phiên mã FOXA2, được ức chế qua insulin-phosphatidylinositol-3-kinase / Akt, và được gây ra bởi tín hiệu glucagon-cAMP-p300 (Arias và cộng sự, 1995; Hegardt, 1999; Quant et al. , 1990; Thumelin và cộng sự, 1993; von Meyenn và cộng sự, 2013; Wolfrum và cộng sự, 2004; Wolfrum và cộng sự, 2003). PPAR? (Rodriguez và cộng sự, 1994) cùng với mục tiêu của nó, FGF21 (Badman và cộng sự, 2007) cũng gây ra phiên mã Hmgcs2 trong gan khi đói hoặc áp dụng chế độ ăn ketogenic (Badman và cộng sự, 2007; Inagaki và cộng sự, 2007 ). Cảm ứng của PPAR? có thể xảy ra trước khi chuyển từ sinh lý bào thai sang sơ sinh, trong khi kích hoạt FGF21 có thể được ưa chuộng trong giai đoạn đầu sơ sinh thông qua sự ức chế histone deacetylase qua trung gian? OHB (HDAC) -3 (Rando và cộng sự, 2016). mTORC1 (đối tượng động vật có vú của phức hợp rapamycin 1) ức chế PPAR phụ thuộc? Hoạt động phiên mã cũng là cơ quan điều hòa chính của sự biểu hiện gen Hmgcs2 (Sengupta và cộng sự, 2010), và PER2 ở gan, một bộ dao động sinh học chính, gián tiếp điều chỉnh biểu hiện Hmgcs2 (Chavan và cộng sự, 2016). Các quan sát gần đây chỉ ra rằng interleukin-6 gây ra khối u ngoài gan làm suy giảm sự tạo ceton thông qua PPAR? đàn áp (Flint và cộng sự, 2016).
Hoạt tính của enzyme HMGCS2 được điều chỉnh thông qua nhiều PTM. HMGCS2 phosphoryl hóa serine tăng cường hoạt tính của nó trong ống nghiệm (Grimsrud et al., 2012). Hoạt tính của HMGCS2 bị ức chế bởi succinyl-CoA và lysine succinylation (Arias và cộng sự, 1995; Hegardt, 1999; Lowe và Tubbs, 1985; Quant và cộng sự, 1990; Rardin và cộng sự, 2013; Reed và cộng sự, 1975; Thumelin và cộng sự, 1993). Succinylation của HMGCS2, HMGCL, và BDH1 dư lượng lysine trong ty thể gan là mục tiêu của NAD + phụ thuộc deacylase sirtuin 5 (SIRT5) (Rardin et al., 2013). Hoạt tính HMGCS2 cũng được tăng cường bằng cách sử dụng lysine deacetylation của SIRT3, và có thể nhiễu xuyên âm giữa acetyl hóa và succinyl hóa điều chỉnh hoạt động HMGCS2 (Rardin et al., 2013; Shimazu và cộng sự, 2013). Mặc dù khả năng của các PTM này để điều chỉnh HMGCS2 Km và Vmax, các biến động của các PTM này vẫn chưa được lập bản đồ cẩn thận và chưa được xác nhận là trình điều khiển cơ học của ketogenesis in vivo.
SCOT được thể hiện trong tất cả các tế bào động vật có vú chứa ti thể, ngoại trừ các tế bào gan. Tầm quan trọng của hoạt động SCOT và ketolysis đã được chứng minh ở chuột SCOT-KO, thể hiện sự mất cân bằng thống nhất do hạ đường huyết tăng huyết áp trong 48h sau khi sinh (Cotter và cộng sự, 2011). Mất mô SCOT cụ thể trong tế bào thần kinh hoặc tế bào cơ xương gây ra bất thường trao đổi chất trong quá trình đói nhưng không gây tử vong (Cotter et al., 2013b). Ở người, sự thiếu hụt SCOT thể hiện sớm trong cuộc sống với tình trạng nhiễm ceton acid nặng, gây hôn mê, nôn mửa và hôn mê (Berry và cộng sự, 2001; Fukao và cộng sự, 2000; Kassovska-Bratinova và cộng sự, 1996; Niezen-Koning et al. , 1997; Saudubray và cộng sự, 1987; Snyderman và cộng sự, 1998; Tildon và Cornblath, 1972). Tương đối ít được biết đến ở cấp độ tế bào về gen điều khiển biểu hiện protein và gen SCOT. Oxct1 mRNA biểu hiện và protein SCOT và hoạt động được giảm trong trạng thái ketotic, có thể thông qua các cơ chế phụ thuộc PPAR (Fenselau và Wallis, 1974; Fenselau và Wallis, 1976; Grinblat et al., 1986; Okuda và cộng sự, 1991; Turko et al ., 2001; Wentz và cộng sự, 2010). Trong nhiễm ketoacidosis tiểu đường, sự không phù hợp giữa ketogenesis gan và quá trình oxy hóa ngoài da trở nên trầm trọng hơn do suy giảm hoạt động của SCOT. Sự biểu hiện quá mức của chất vận chuyển glucose độc lập với insulin (GLUT1 / SLC2A1) trong các tế bào cơ tim cũng ức chế sự biểu hiện gen Oxct1 và làm giảm quá trình oxy hóa thiết bị đầu cuối xeton trong trạng thái không ketotic (Yan et al., 2009). Trong gan, sự đa dạng của Oxct1 mRNA bị ức chế bởi microRNA-122 và methyl hóa histone H3K27me3 rõ ràng trong quá trình chuyển từ thai nhi sang giai đoạn sơ sinh (Thorrez et al., 2011). Tuy nhiên, sự ức chế biểu hiện Oxct1 ở gan trong giai đoạn hậu sản chủ yếu là do sự di tản của các tế bào tạo máu biểu hiện Oxct1 từ gan, thay vì mất biểu hiện Oxct1 trước đây trong các tế bào gan khác biệt. Trong thực tế, sự biểu hiện của protein Oxct1 mRNA và SCOT trong các tế bào gan khác biệt là rất thấp (Orii và cộng sự, 2008).
SCOT cũng được quy định bởi các PTM. Enzyme này bị siêu acetyl hóa trong não của chuột SIRT3 KO, cũng cho thấy khả năng sản xuất acetyl-CoA phụ thuộc AcAc giảm dần (Dittenhafer-Reed et al., 2015). Quá trình nitrat hóa không do enzym của dư lượng tyrosine của SCOT cũng làm giảm hoạt tính của nó, điều này đã được báo cáo trong tim của các mô hình chuột mắc bệnh tiểu đường khác nhau (Marcondes và cộng sự, 2001; Turko và cộng sự, 2001; Wang và cộng sự, 2010a). Ngược lại, nitrat dư lượng tryptophan làm tăng hoạt tính của SCOT (Br g re et al., 2010; Rebrin et al., 2007). Có thể tồn tại các cơ chế phân tử của quá trình nitrat hóa hoặc khử nitơ cụ thể theo dư lượng được thiết kế để điều chỉnh hoạt động của SCOT và cần được làm sáng tỏ.
Những tranh cãi trong sinh sản ngoài da
Ở động vật có vú, cơ quan sinh xeton chính là gan, và chỉ tế bào gan và tế bào biểu mô ruột biểu hiện phong phú dạng đồng dạng ty thể của HMGCS2 (Cotter và cộng sự, 2013a; Cotter và cộng sự, 2014; McGarry và Foster, 1980; Robinson và Williamson, 1980) . Quá trình lên men vi khuẩn kỵ khí tạo ra các polysaccharid phức tạp tạo ra butyrate, được hấp thụ bởi các tế bào màu ở động vật có vú để oxy hóa giai đoạn cuối hoặc tạo ketogenesis (Cherbuy và cộng sự, 1995), có thể đóng một vai trò trong sự khác biệt của tế bào màu (Wang và cộng sự, 2016). Không bao gồm các tế bào biểu mô ruột và tế bào gan, HMGCS2 gần như không có trong hầu hết các tế bào động vật có vú khác, nhưng triển vọng hình thành ketogenesis ngoài gan đã được đặt ra ở các tế bào khối u, tế bào hình sao của hệ thần kinh trung ương, thận, tuyến tụy? tế bào, biểu mô sắc tố võng mạc (RPE), và thậm chí trong cơ xương (Adijanto và cộng sự, 2014; Avogaro và cộng sự, 1992; El Azzouny và cộng sự, 2016; Grabacka và cộng sự, 2016; Kang và cộng sự, 2015 ; Le Foll và cộng sự, 2014; Nonaka và cộng sự, 2016; Takagi và cộng sự, 2016a; Thevenet và cộng sự, 2016; Zhang và cộng sự, 2011). HMGCS2 ngoài tử cung đã được quan sát thấy trong các mô thiếu khả năng tạo ketogenic thuần (Cook và cộng sự, 2016; Wentz và cộng sự, 2010), và HMGCS2 thể hiện các hoạt động trăng sáng không phụ thuộc vào quá trình tạo ketogenesis, bao gồm cả trong nhân tế bào (Chen và cộng sự. , 2016; Kostiuk và cộng sự, 2010; Meertens và cộng sự, 1998).
Bất kỳ mô ngoài gan nào oxy hóa các thể xeton cũng có khả năng tích tụ các thể xeton thông qua cơ chế độc lập HMGCS2 (Hình 2A). Tuy nhiên, không có mô ngoài gan nào mà ở đó nồng độ ceton trong cơ thể ở trạng thái ổn định vượt quá nồng độ trong tuần hoàn (Cotter và cộng sự, 2011; Cotter và cộng sự, 2013b; Harrison và Long, 1940), nhấn mạnh rằng các thể xeton được vận chuyển xuống một gradient nồng độ thông qua cơ chế phụ thuộc MCT1 / 2. Một cơ chế tạo xeton ngoài gan rõ ràng có thể thực sự phản ánh sự suy giảm tương đối của quá trình oxy hóa xeton. Các giải thích tiềm năng bổ sung nằm trong lĩnh vực hình thành cơ thể xeton. Đầu tiên, quá trình tạo xeton de novo có thể xảy ra thông qua hoạt động enzym thuận nghịch của thiolase và SCOT (Weidemann và Krebs, 1969). Khi nồng độ acetyl-CoA tương đối cao, các phản ứng thường chịu trách nhiệm về quá trình oxy hóa AcAc hoạt động theo chiều ngược lại (GOLDMAN, 1954). Cơ chế thứ hai xảy ra khi các chất trung gian có nguồn gốc?-Oxy hóa tích tụ do tắc nghẽn chu trình TCA, AcAc-CoA được chuyển đổi thành l-? OHB-CoA thông qua một phản ứng được xúc tác bởi 3-hydroxyacyl-CoA dehydrogenase của ty thể, và thêm nữa bởi 3-hydroxybutyryl CoA deacylase thành l- OHB, không thể phân biệt bằng khối phổ hoặc phổ cộng hưởng với đồng phân đối quang sinh lý d-? OHB (Reed và Ozand, 1980). l-? OHB có thể được phân biệt bằng sắc ký hoặc enzym với d-? OHB, và có trong các mô ngoài gan, nhưng không có trong gan hoặc máu (Hsu và cộng sự, 2011). Quá trình tạo xeton ở gan chỉ tạo ra d-? OHB, chất đồng phân đối quang duy nhất là chất nền BDH (Ito và cộng sự, 1984; Lincoln và cộng sự, 1987; Reed và Ozand, 1980; Scofield và cộng sự, 1982; Scofield và cộng sự, Năm 1982). Cơ chế thứ ba không phụ thuộc vào HMGCS2 tạo ra d- OHB thông qua quá trình dị hóa axit amin, đặc biệt là của leucine và lysine. Cơ chế thứ tư chỉ rõ ràng bởi vì nó là do một tạo tác gắn nhãn và do đó được gọi là sự phát sinh giả. Hiện tượng này là do sự đảo ngược của phản ứng SCOT và thiolase, và có thể gây ra đánh giá quá cao sự luân chuyển của cơ thể xeton do sự pha loãng đồng vị của chất đánh dấu cơ thể xeton trong mô ngoài gan (Des Rosiers et al., 1990; Fink et al., 1988) . Tuy nhiên, sự phát sinh giả có thể không đáng kể trong hầu hết các bối cảnh (Bailey và cộng sự, 1990; Keller và cộng sự, 1978). Một giản đồ (Hình 2A) chỉ ra một cách tiếp cận hữu ích để áp dụng trong khi xem xét nồng độ xeton ở trạng thái ổn định trong mô tăng lên.


Thận gần đây đã được chú ý như một cơ quan có khả năng sinh xeton. Ở đại đa số các bang, thận là nơi tiêu thụ ròng các thể xeton có nguồn gốc từ gan, bài tiết hoặc tái hấp thu các thể xeton từ máu, và thận nói chung không phải là cơ quan tạo hoặc tập trung cơ thể xeton thuần (Robinson và Williamson, 1980). Các tác giả của một nghiên cứu cổ điển kết luận rằng lượng ketogenesis tối thiểu ở thận được định lượng trong một hệ thống thí nghiệm nhân tạo không có liên quan về mặt sinh lý học (Weidemann và Krebs, 1969). Gần đây, sự tạo ceton ở thận đã được suy ra trong các mô hình chuột mắc bệnh tiểu đường và thiếu máu tự động, nhưng có nhiều khả năng sự thay đổi đa cơ quan trong cân bằng nội môi chuyển hóa làm thay đổi quá trình chuyển hóa xeton tích hợp thông qua đầu vào trên nhiều cơ quan (Takagi et al., 2016a; Takagi et al., 2016b; Zhang và cộng sự, 2011). Một công bố gần đây đề xuất sự tạo ceton ở thận như một cơ chế bảo vệ chống lại tổn thương do thiếu máu cục bộ tái tưới máu ở thận (Tran et al., 2016). Nồng độ ở trạng thái ổn định tuyệt đối của? OHB từ chiết xuất của mô thận chuột được báo cáo ở ~ 4 12 mM. Để kiểm tra xem điều này có thể nghe được hay không, chúng tôi đã định lượng nồng độ OHB trong dịch chiết thận từ những con chuột được cho ăn và nhịn ăn trong 24 giờ. Nồng độ OHB trong huyết thanh tăng từ ~ 100 M lên 2 mM khi nhịn đói 24h (Hình 2B), trong khi ở trạng thái ổn định ở thận, nồng độ OHB xấp xỉ 100 M ở trạng thái được cho ăn và chỉ 1 mM ở trạng thái nhịn ăn 24h (Hình. 2C E), những quan sát phù hợp với nồng độ đã được định lượng hơn 45 năm trước (Hems và Brosnan, 1970). Vẫn có thể là ở trạng thái xeton, các thể xeton có nguồn gốc từ gan có thể được tái bảo vệ, nhưng bằng chứng về sự tạo xeton ở thận cần được chứng minh thêm. Bằng chứng thuyết phục hỗ trợ sự hình thành ketogenesis ngoài gan thực sự đã được trình bày trong RPE (Adijanto và cộng sự, 2014). Sự biến đổi trao đổi chất hấp dẫn này được đề xuất là có khả năng cho phép các xeton có nguồn gốc từ RPE chảy đến tế bào cảm thụ ánh sáng hoặc tế bào M ller, có thể hỗ trợ tái tạo phân đoạn bên ngoài cơ quan thụ cảm ánh sáng.
? OHB với tư cách là Người hòa giải tín hiệu
Mặc dù chúng rất giàu năng lượng, các cơ thể xeton có vai trò truyền tín hiệu non-canonical khiêu khích trong việc cân bằng nội môi tế bào (Hình 3) (Newman và Verdin, 2014; Rojas-Morales và cộng sự, 2016). Ví dụ,? OHB ức chế HDAC loại I, làm tăng quá trình acetyl hóa histone và do đó gây ra sự biểu hiện của các gen ngăn chặn stress oxy hóa (Shimazu et al., 2013). Bản thân OHB là một chất điều chỉnh cộng hóa trị histone tại dư lượng lysine trong gan của chuột mắc bệnh tiểu đường nhịn ăn hoặc do streptozotocin gây ra (Xie và cộng sự, 2016) (cũng xem bên dưới, Sự tích hợp của chuyển hóa cơ thể xeton, biến đổi sau dịch mã và sinh lý tế bào, và Cơ thể xeton, stress oxy hóa và bảo vệ thần kinh).

OHB cũng là một tác nhân thông qua các thụ thể kết hợp với protein G. Thông qua các cơ chế phân tử không rõ ràng, nó ngăn chặn hoạt động của hệ thần kinh giao cảm và làm giảm tổng tiêu hao năng lượng và nhịp tim bằng cách ức chế tín hiệu axit béo chuỗi ngắn thông qua thụ thể kết hợp với protein G 41 (GPR41) (Kimura và cộng sự, 2011). Một trong những hiệu ứng tín hiệu được nghiên cứu nhiều nhất của? OHB tiến hành thông qua GPR109A (còn được gọi là HCAR2), một thành viên của phân họ GPCR axit hydrocacboxylic được biểu hiện trong các mô mỡ (trắng và nâu) (Tunaru và cộng sự, 2003), và tế bào miễn dịch (Ahmed và cộng sự, 2009). OHB là phối tử nội sinh duy nhất được biết đến của thụ thể GPR109A (EC50 ~ 770 M) được kích hoạt bởi d-? OHB, l-? OHB và butyrate, nhưng không phải AcAc (Taggart và cộng sự, 2005). Ngưỡng nồng độ cao để kích hoạt GPR109A đạt được thông qua việc tuân thủ chế độ ăn ketogenic, bỏ đói hoặc trong quá trình nhiễm toan ceton, dẫn đến ức chế phân giải lipid mô mỡ. Tác dụng chống phân giải mỡ của GPR109A diễn ra thông qua việc ức chế adenylyl cyclase và giảm cAMP, ức chế lipase triglyceride nhạy cảm với hormone (Ahmed và cộng sự, 2009; Tunaru và cộng sự, 2003). Điều này tạo ra một vòng phản hồi tiêu cực, trong đó ketosis đặt một phanh điều hòa trên quá trình tạo xeton bằng cách giảm giải phóng các axit béo không được este hóa từ các tế bào mỡ (Ahmed và cộng sự, 2009; Taggart và cộng sự, 2005), một hiệu ứng có thể được cân bằng bằng cách ổ giao cảm kích thích phân giải lipid. Niacin (vitamin B3, axit nicotinic) là phối tử mạnh (EC50 ~ 0.1 M) cho GRP109A, được sử dụng hiệu quả trong nhiều thập kỷ cho chứng rối loạn lipid máu (Benyo và cộng sự, 2005; Benyo và cộng sự, 2006; Fabbrini và cộng sự, 2010a; Lukasova và cộng sự, 2011; Tunaru và cộng sự, 2003). Trong khi niacin tăng cường vận chuyển ngược lại cholesterol trong đại thực bào và làm giảm các tổn thương xơ vữa động mạch (Lukasova và cộng sự, 2011), tác động của? OHB đối với các tổn thương xơ vữa vẫn chưa được biết rõ. Mặc dù thụ thể GPR109A có vai trò bảo vệ và tồn tại các mối liên hệ hấp dẫn giữa việc sử dụng chế độ ăn ketogenic trong đột quỵ và các bệnh thoái hóa thần kinh (Fu và cộng sự, 2015; Rahman và cộng sự, 2014), vai trò bảo vệ của? OHB thông qua GPR109A chưa được chứng minh trên cơ thể sống .
Cuối cùng,? OHB có thể ảnh hưởng đến sự thèm ăn và cảm giác no. Một phân tích tổng hợp các nghiên cứu đo lường tác động của chế độ ăn ketogenic và rất ít năng lượng đã kết luận rằng những người tham gia ăn những chế độ ăn này có cảm giác no cao hơn so với chế độ ăn đối chứng (Gibson và cộng sự, 2015). Tuy nhiên, một lời giải thích hợp lý cho tác động này là các yếu tố chuyển hóa hoặc nội tiết tố bổ sung có thể điều chỉnh sự thèm ăn. Ví dụ, những con chuột được duy trì theo chế độ ăn ketogenic cho loài gặm nhấm biểu hiện mức tiêu hao năng lượng tăng lên so với những con chuột được cho ăn đối chứng chow, mặc dù lượng calo tương tự và leptin lưu hành hoặc các gen peptide điều chỉnh hành vi cho ăn không bị thay đổi (Kennedy và cộng sự, 2007). Trong số các cơ chế được đề xuất đề xuất ức chế sự thèm ăn bằng? OHB bao gồm cả tín hiệu và quá trình oxy hóa (Laeger et al., 2010). Tế bào gan xóa đặc hiệu gen nhịp sinh học (Per2) và nghiên cứu kết tủa miễn dịch nhiễm sắc cho thấy PER2 trực tiếp kích hoạt gen Cpt1a, và gián tiếp điều chỉnh Hmgcs2, dẫn đến suy giảm ketosis ở chuột bị loại Per2 (Chavan et al., 2016). Những con chuột này có biểu hiện suy giảm khả năng dự đoán thức ăn, điều này được phục hồi một phần bằng cách sử dụng? OHB toàn thân. Các nghiên cứu trong tương lai sẽ là cần thiết để xác nhận hệ thống thần kinh trung ương là mục tiêu trực tiếp của OHB, và liệu quá trình oxy hóa xeton có cần thiết cho các tác động quan sát được hay không hoặc liệu một cơ chế tín hiệu khác có liên quan hay không. Các nhà nghiên cứu khác đã đưa ra khả năng hình thành xeton có nguồn gốc tế bào hình sao cục bộ trong vùng dưới đồi não thất như một cơ quan điều chỉnh lượng thức ăn, nhưng những quan sát sơ bộ này cũng sẽ có lợi từ các đánh giá dựa trên di truyền và thông lượng (Le Foll và cộng sự, 2014). Mối quan hệ giữa ketosis và thiếu hụt chất dinh dưỡng vẫn được quan tâm vì đói và no là những yếu tố quan trọng trong nỗ lực giảm cân thất bại.
Tích hợp chuyển hóa cơ thể Ketone, sửa đổi sau phiên dịch và sinh lý tế bào
Các cơ thể Ketone đóng góp vào các bể chứa acetyl-CoA, một chất trung gian quan trọng thể hiện vai trò nổi bật trong chuyển hóa tế bào (Pietrocola et al., 2015). Một vai trò của acetyl-CoA là đóng vai trò là chất nền cho acetyl hóa, điều chỉnh cộng hóa trị histone được xúc tác bằng enzyme (Choudhary et al., 2014; Dutta et al., 2016; Fan et al., 2015; Menzies et al. ). Một số lượng lớn các protein ty thể được acetyl hóa tự động, nhiều trong số đó có thể xảy ra thông qua các cơ chế không enzyme, cũng đã xuất hiện từ các nghiên cứu proteomics tính toán (Dittenhafer-Reed et al., 2016; Hebert et al., 2015; Rardin et al. ; Shimemo và cộng sự, 2013). Lysine deacetylase sử dụng một đồng yếu tố kẽm (ví dụ, HDAC nucleocytosolic) hoặc NAD + làm chất nền (sirtuins, SIRTs) (Choudhary et al., 2013; Menzies et al., 2010). Acetylproteome phục vụ như là cả cảm biến và effector của tổng số di động acetyl-CoA hồ bơi, như thao tác sinh lý và di truyền mỗi kết quả trong các biến thể không enzyme toàn cầu của acetylation (Weinert et al., 2014). Khi các chất chuyển hóa nội bào đóng vai trò là bộ điều biến của quá trình acetyl hóa dư lượng lysine, điều quan trọng là phải xem xét vai trò của các cơ quan xeton, có sự phong phú rất năng động.
OHB là một chất điều chỉnh biểu sinh thông qua ít nhất hai cơ chế. Nồng độ OHB tăng lên do nhịn ăn, hạn chế calo, sử dụng trực tiếp hoặc tập thể dục kéo dài gây ra ức chế HDAC hoặc kích hoạt histone acetyltransferase (Marosi và cộng sự, 2016; Sleiman và cộng sự, 2016) hoặc do căng thẳng oxy hóa (Shimazu và cộng sự, 2013) . Sự ức chế OHB của HDAC3 có thể điều chỉnh sinh lý trao đổi chất ở trẻ sơ sinh (Rando và cộng sự, 2016). OHB tự điều chỉnh trực tiếp dư lượng histone lysine (Xie và cộng sự, 2016). Nhịn ăn kéo dài, hoặc nhiễm toan ceton do đái tháo đường gây ra bởi steptozotocin làm tăng histone? -Hydroxybutyrylation. Mặc dù số lượng các vị trí lysine? -Hydroxybutyryl hóa và acetyl hóa là tương đương nhau, nhưng histone? -Hydroxybutyrylation lớn hơn so với acetyl hóa được quan sát thấy. Các gen khác biệt bị tác động bởi quá trình phản ứng histone lysine? -Hydroxybutyryl hóa, so với quá trình acetyl hóa hoặc methyl hóa, cho thấy các chức năng tế bào khác nhau. Người ta chưa biết liệu? -Hydroxybutyryl hóa là tự phát hay enzym, nhưng mở rộng phạm vi cơ chế thông qua các thể xeton ảnh hưởng động đến quá trình phiên mã.
Các sự kiện tái lập trình tế bào cần thiết trong quá trình hạn chế calo và thiếu hụt chất dinh dưỡng có thể được điều hòa trong quá trình khử oxy hóa ty thể phụ thuộc SIRT3 và SIRT5 và quá trình khử chất béo tương ứng, điều chỉnh các protein ketogenic và ketolytic ở mức độ sau dịch mã trong gan và các mô ngoài gan (Dittenhafer-Reed et al., 2015; Hebert và cộng sự, 2013; Rardin và cộng sự, 2013; Shimazu và cộng sự, 2010). Mặc dù so sánh phân vị giữa các vị trí bị chiếm đóng không nhất thiết liên quan trực tiếp đến sự thay đổi trong dòng trao đổi chất, nhưng quá trình acetyl hóa ty thể là động lực và có thể được thúc đẩy bởi nồng độ acetyl-CoA hoặc độ pH của ty thể, chứ không phải là acetyltransferase của enzym (Wagner và Payne, 2013). SIRT3 và SIRT5 điều chỉnh hoạt động của các enzym chuyển hóa cơ thể xeton đặt ra câu hỏi về vai trò tương hỗ của xeton trong việc tạo ra acetylproteome, succinylproteome và các mục tiêu tế bào động khác. Thật vậy, khi các biến thể của quá trình tạo xeton phản ánh nồng độ NAD +, việc sản xuất xeton và sự phong phú có thể điều chỉnh hoạt động của sirtuin, do đó ảnh hưởng đến tổng lượng acetyl-CoA / succinyl-CoA, acylproteome, và do đó sinh lý học ty thể và tế bào. ? -hydroxybutyryl hóa dư lượng enzyme lysine có thể thêm một lớp khác để tái lập trình tế bào. Trong các mô ngoài gan, quá trình oxy hóa cơ thể xeton có thể kích thích những thay đổi tương tự trong cân bằng nội môi của tế bào. Trong khi việc phân chia vùng chứa acetyl-CoA được điều chỉnh cao và điều phối một loạt các thay đổi tế bào, khả năng của các thể xeton trực tiếp định hình cả nồng độ acetyl-CoA trong ty thể và tế bào chất đòi hỏi phải được làm sáng tỏ (Chen và cộng sự, 2012; Corbet và cộng sự, 2016; Pougovkina và cộng sự, 2014; Schwer và cộng sự, 2009; Wellen và Thompson, 2012). Bởi vì nồng độ acetyl-CoA được điều chỉnh chặt chẽ và acetyl-CoA là chất không thấm qua màng, điều quan trọng là phải xem xét các cơ chế điều khiển điều phối cân bằng nội môi acetyl-CoA, bao gồm tốc độ sản xuất và quá trình oxy hóa cuối trong chu trình TCA, chuyển đổi thành thể xeton, ty thể. chảy ra qua carnitine acetyltransferase (CrAT), hoặc acetyl-CoA xuất sang cytosol sau khi chuyển đổi thành citrate và được giải phóng bởi ATP citrate lyase (ACLY). Vai trò quan trọng của các cơ chế thứ hai này trong acetylproteome tế bào và cân bằng nội môi đòi hỏi sự hiểu biết phù hợp về vai trò của quá trình tạo xeton và quá trình oxy hóa xeton (Das et al., 2015; McDonnell et al., 2016; Moussaieff et al., 2015; Overmyer et al., 2015; Seiler và cộng sự, 2014; Seiler và cộng sự, 2015; Wellen và cộng sự, 2009; Wellen và Thompson, 2012). Công nghệ hội tụ trong chuyển hóa và acylproteomics trong việc thiết lập các mô hình thao tác di truyền sẽ được yêu cầu để xác định các mục tiêu và kết quả.
Phản ứng chống và chống viêm đối với các cơ quan Ketone
Các cơ quan xeton và xeton điều chỉnh tình trạng viêm và chức năng tế bào miễn dịch, nhưng các cơ chế khác nhau và thậm chí khác nhau đã được đề xuất. Thiếu hụt chất dinh dưỡng kéo dài làm giảm viêm (Youm và cộng sự, 2015), nhưng ketosis mãn tính của bệnh tiểu đường loại 1 là trạng thái tiền viêm (Jain và cộng sự, 2002; Kanikarla-Marie và Jain, 2015; Kurepa và cộng sự, 2012 ). Vai trò tín hiệu dựa trên cơ chế đối với? OHB trong viêm xuất hiện do nhiều tế bào của hệ thống miễn dịch, bao gồm đại thực bào hoặc bạch cầu đơn nhân, biểu hiện rất nhiều GPR109A. Trong khi? OHB thực hiện phản ứng chống viêm chủ yếu (Fu và cộng sự, 2014; Gambhir và cộng sự, 2012; Rahman và cộng sự, 2014; Youm và cộng sự, 2015), nồng độ cao của các thể xeton, đặc biệt là AcAc, có thể kích hoạt phản ứng tiền viêm (Jain và cộng sự, 2002; Kanikarla-Marie và Jain, 2015; Kurepa và cộng sự, 2012).
Vai trò chống viêm của phối tử GPR109A trong xơ vữa động mạch, béo phì, bệnh viêm ruột, bệnh thần kinh và ung thư đã được xem xét (Graff và cộng sự, 2016). Biểu hiện GPR109A được tăng cường trong các tế bào RPE của các mô hình đái tháo đường, bệnh nhân đái tháo đường ở người (Gambhir và cộng sự, 2012), và trong bệnh tiểu đường trong quá trình thoái hóa thần kinh (Fu và cộng sự, 2014). Tác dụng chống viêm của? OHB được tăng cường bởi sự biểu hiện quá mức của GPR109A trong các tế bào RPE và bị thay thế bởi sự ức chế dược lý hoặc loại bỏ di truyền của GPR109A (Gambhir và cộng sự, 2012). OHB và axit nicotinic ngoại sinh (Taggart và cộng sự, 2005), cả hai đều tạo ra tác dụng chống viêm trong TNF? hoặc viêm do LPS bằng cách giảm mức độ protein chống viêm (iNOS, COX-2), hoặc các cytokine tiết ra (TNF ?, IL-1 ?, IL-6, CCL2 / MCP-1), một phần thông qua việc ức chế NF -? B chuyển vị (Fu và cộng sự, 2014; Gambhir và cộng sự, 2012). OHB làm giảm căng thẳng ER và sự phát sinh NLRP3, kích hoạt phản ứng chống stress chống oxy hóa (Bae và cộng sự, 2016; Youm và cộng sự, 2015). Tuy nhiên, trong viêm thoái hóa thần kinh, bảo vệ qua trung gian OHB phụ thuộc GPR109A không liên quan đến các chất trung gian gây viêm như tín hiệu con đường MAPK (ví dụ, ERK, JNK, p38) (Fu và cộng sự, 2014), nhưng có thể yêu cầu PGD1 phụ thuộc COX-2 sản xuất (Rahman và cộng sự, 2014). Điều hấp dẫn là đại thực bào GPR109A được yêu cầu để phát huy tác dụng bảo vệ thần kinh trong mô hình đột quỵ do thiếu máu cục bộ (Rahman và cộng sự, 2014), nhưng khả năng của? OHB để ức chế NLRP3 trong đại thực bào có nguồn gốc từ tủy xương là không phụ thuộc vào GPR109A (Youm và cộng sự ., 2015). Mặc dù hầu hết các nghiên cứu liên kết? OHB với tác dụng chống viêm, OHB có thể gây viêm và làm tăng các dấu hiệu của quá trình peroxy hóa lipid trong tế bào gan bê (Shi và cộng sự, 2014). Do đó, tác dụng chống và chống viêm của? OHB có thể phụ thuộc vào loại tế bào, nồng độ? OHB, thời gian tiếp xúc và sự hiện diện hay không có của chất đồng điều chế.
Không giống như OHB, AcAc có thể kích hoạt tín hiệu tiền viêm. AcAc tăng cao, đặc biệt là với nồng độ glucose cao, làm tăng cường tổn thương tế bào nội mô thông qua cơ chế phụ thuộc stress oxy hóa / NADPH oxidase (Kanikarla-Marie và Jain, 2015). Nồng độ AcAc cao trong dây rốn của các bà mẹ bị tiểu đường có tương quan với tốc độ oxy hóa protein và nồng độ MCP-1 cao hơn (Kurepa và cộng sự, 2012). AcAc cao ở bệnh nhân đái tháo đường có tương quan với TNF không? biểu hiện (Jain và cộng sự, 2002), và AcAc, nhưng không phải? OHB, gây ra TNF ?, biểu hiện MCP-1, tích tụ ROS và giảm mức cAMP trong tế bào bạch cầu đơn nhân U937 (Jain và cộng sự, 2002; Kurepa và cộng sự ., 2012).
Hiện tượng truyền tín hiệu phụ thuộc vào cơ thể xeton thường chỉ được kích hoạt khi nồng độ xeton trong cơ thể cao (> 5 mM), và trong trường hợp nhiều nghiên cứu liên kết xeton với tác dụng chống viêm hoặc chống viêm, thông qua các cơ chế không rõ ràng. Ngoài ra, do tác động trái ngược nhau của? OHB so với AcAc đối với phản ứng viêm và khả năng của tỷ lệ AcAc /? OHB ảnh hưởng đến điện thế oxy hóa khử của ty thể, các thí nghiệm tốt nhất đánh giá vai trò của thể xeton đối với kiểu hình tế bào so sánh tác dụng của AcAc và? OHB ở các tỷ lệ khác nhau, và ở các nồng độ tích lũy khác nhau [ví dụ, (Saito và cộng sự, 2016)]. Cuối cùng, AcAc chỉ có thể được mua thương mại dưới dạng muối liti hoặc như một este etylic cần thủy phân bazơ trước khi sử dụng. Cation liti độc lập tạo ra các tầng dẫn truyền tín hiệu (Manji và cộng sự, 1995), và anion AcAc là không bền. Cuối cùng, các nghiên cứu sử dụng racemic d / l-? OHB có thể bị nhầm lẫn, vì chỉ đồng phân lập thể d-? OHB mới có thể bị oxy hóa thành AcAc, nhưng d-? OHB và l-? OHB mỗi người có thể phát tín hiệu qua GPR109A, ức chế vi khuẩn NLRP3, và đóng vai trò là chất nền tạo mỡ.
Ketone Bodies, Stress oxy hóa và bảo vệ thần kinh
Ứng suất oxy hóa thường được định nghĩa là trạng thái trong đó ROS được xuất hiện quá mức, do sản xuất quá mức và / hoặc sự đào thải bị suy giảm. Vai trò chống oxy hóa và giảm stress oxy hóa của các thể xeton đã được mô tả rộng rãi cả in vitro và in vivo, đặc biệt trong bối cảnh bảo vệ thần kinh. Vì hầu hết các tế bào thần kinh không tạo ra phốt phát năng lượng cao một cách hiệu quả từ các axit béo nhưng lại oxy hóa các thể xeton khi thiếu cacbohydrat, nên tác dụng bảo vệ thần kinh của các thể xeton là đặc biệt quan trọng (Cahill GF Jr, 2006; Edmond và cộng sự, 1987; Yang và cộng sự, 1987). Trong các mô hình stress oxy hóa, cảm ứng BDH1 và ức chế SCOT gợi ý rằng quá trình chuyển hóa xeton trong cơ thể có thể được lập trình lại để duy trì các yêu cầu về tín hiệu tế bào, điện thế oxy hóa khử hoặc trao đổi chất đa dạng (Nagao et al., 2016; Tieu et al., 2003).
Các thể xeton làm giảm mức độ tổn thương tế bào, tổn thương, tử vong và giảm quá trình chết rụng ở tế bào thần kinh và tế bào cơ tim (Haces và cộng sự, 2008; Maalouf và cộng sự, 2007; Nagao và cộng sự, 2016; Tieu và cộng sự, 2003). Các cơ chế được mời rất đa dạng và không phải lúc nào cũng liên quan tuyến tính với nồng độ. Nồng độ milimolar thấp của (d hoặc l) -? OHB quét ROS (hydroxyl anion), trong khi AcAc quét nhiều loài ROS, nhưng chỉ ở nồng độ vượt quá phạm vi sinh lý (IC50 20 67 mM) (Haces et al., 2008) . Ngược lại, ảnh hưởng có lợi đến tiềm năng oxy hóa khử của chuỗi vận chuyển electron là một cơ chế thường được liên kết với d- OHB. Trong khi cả ba thể xeton (d / l-? OHB và AcAc) làm giảm quá trình chết của tế bào thần kinh và sự tích tụ ROS được kích hoạt bởi sự ức chế hóa học của quá trình đường phân, chỉ có d-? OHB và AcAc ngăn chặn sự suy giảm ATP của tế bào thần kinh. Ngược lại, trong mô hình in vivo hạ đường huyết, (d hoặc l) -? OHB, nhưng không phải AcAc ngăn chặn quá trình peroxy hóa lipid ở vùng hải mã (Haces và cộng sự, 2008; Maalouf và cộng sự, 2007; Marosi và cộng sự, 2016; Murphy, 2009 ; Tieu và cộng sự, 2003). Các nghiên cứu in vivo đối với những con chuột được cho ăn chế độ ăn ketogenic (87% kcal chất béo và 13% protein) cho thấy sự biến đổi cơ chế thần kinh của khả năng chống oxy hóa (Ziegler và cộng sự, 2003), trong đó những thay đổi sâu sắc nhất được quan sát thấy ở hải mã, với sự gia tăng glutathione peroxidase và tổng số khả năng chống oxy hóa.
Chế độ ăn ketogenic, các este xeton (cũng xem Sử dụng điều trị của chế độ ăn ketogenic và các cơ thể xeton ngoại sinh), hoặc? Quản lý OHB có tác dụng bảo vệ thần kinh trong các mô hình đột quỵ do thiếu máu cục bộ (Rahman và cộng sự, 2014); Bệnh Parkinson (Tieu và cộng sự, 2003); co giật do nhiễm độc oxy của hệ thần kinh trung ương (D'Agostino và cộng sự, 2013); co thắt động kinh (Yum và cộng sự, 2015); bệnh cơ não ty thể, nhiễm toan lactic và hội chứng các cơn giống đột quỵ (MELAS) (Frey và cộng sự, 2016) và bệnh Alzheimer (Cunnane và Crawford, 2003; Yin và cộng sự, 2016). Ngược lại, một báo cáo gần đây đã chứng minh bằng chứng mô bệnh học về sự tiến triển thoái hóa thần kinh do chế độ ăn ketogenic ở mô hình chuột chuyển gen về việc sửa chữa DNA ty thể bất thường, mặc dù sự gia tăng quá trình sinh học ty thể và các dấu hiệu chống oxy hóa (Lauritzen và cộng sự, 2016). Các báo cáo mâu thuẫn khác cho thấy rằng việc tiếp xúc với nồng độ xeton cao trong cơ thể gây ra căng thẳng oxy hóa. Liều OHB hoặc AcAc cao gây ra bài tiết oxit nitric, peroxy hóa lipid, giảm biểu hiện của SOD, glutathione peroxidase và catalase trong tế bào gan bê, trong khi ở tế bào gan chuột, sự cảm ứng con đường MAPK được cho là do AcAc chứ không phải? OHB (Abdelmegeed et al., 2004 ; Shi và cộng sự, 2014; Shi và cộng sự, 2016).
Tổng hợp lại, hầu hết các báo cáo đều liên kết? OHB với việc giảm stress oxy hóa, vì việc sử dụng nó ức chế sản xuất ROS / superoxide, ngăn chặn quá trình peroxy hóa lipid và oxy hóa protein, làm tăng mức độ protein chống oxy hóa, và cải thiện hô hấp của ty thể và sản xuất ATP (Abdelmegeed et al., 2004; Haces và cộng sự, 2008; Jain và cộng sự, 1998; Jain và cộng sự, 2002; Kanikarla-Marie và Jain, 2015; Maalouf và cộng sự, 2007; Maalouf và Rho, 2008; Marosi và cộng sự, 2016; Tiêu và cộng sự, 2003; Yin và cộng sự, 2016; Ziegler và cộng sự, 2003). Trong khi AcAc có tương quan trực tiếp hơn? OHB với việc gây ra stress oxy hóa, những tác động này không phải lúc nào cũng dễ dàng được phân tích từ các phản ứng tiền viêm tương lai (Jain và cộng sự, 2002; Kanikarla-Marie và Jain, 2015; Kanikarla-Marie và Jain, 2016). Hơn nữa, điều quan trọng là phải xem xét rằng lợi ích chống oxy hóa rõ ràng mang lại bởi chế độ ăn ketogenic toàn thân có thể không được chuyển hóa bởi chính các cơ quan xeton, và sự bảo vệ thần kinh do cơ thể xeton mang lại có thể không hoàn toàn là do stress oxy hóa. Ví dụ: trong quá trình thiếu hụt glucose, trong một mô hình thiếu hụt glucose ở tế bào thần kinh vỏ não,? OHB kích thích thông lượng tự thực và ngăn chặn sự tích tụ autophagosome, có liên quan đến việc giảm chết tế bào thần kinh (Camberos-Luna và cộng sự, 2016). d-? OHB cũng tạo ra các protein chống oxy hóa kinh điển FOXO3a, SOD, MnSOD và catalase, một cách tiềm năng thông qua ức chế HDAC (Nagao và cộng sự, 2016; Shimazu và cộng sự, 2013).
Bệnh gan nhiễm mỡ không do rượu (NAFLD) và chuyển hóa cơ thể Ketone
NAFLD liên quan đến béo phì và viêm gan nhiễm mỡ không do rượu (NASH) là những nguyên nhân phổ biến nhất của bệnh gan ở các nước phương Tây (Rinella và Sanyal, 2016), và suy gan do NASH là một trong những lý do phổ biến nhất để ghép gan. Trong khi dự trữ dư thừa triacylglycerol trong tế bào gan> 5% trọng lượng gan (NAFL) đơn thuần không gây thoái hóa chức năng gan, thì sự tiến triển thành NAFLD ở người tương quan với tình trạng kháng insulin toàn thân và tăng nguy cơ mắc bệnh tiểu đường loại 2, và có thể góp phần vào cơ chế bệnh sinh của bệnh tim mạch và bệnh thận mãn tính (Fabbrini và cộng sự, 2009; Targher và cộng sự, 2010; Targher và Byrne, 2013). Các cơ chế gây bệnh của NAFLD và NASH vẫn chưa được hiểu đầy đủ nhưng bao gồm các bất thường về chuyển hóa tế bào gan, quá trình tự phân hủy tế bào gan và căng thẳng mạng lưới nội chất, chức năng tế bào miễn dịch gan, viêm mô mỡ và các chất trung gian gây viêm hệ thống (Fabbrini và cộng sự, 2009; Masuoka và Chalasani, 2013 ; Targher và cộng sự, 2010; Yang và cộng sự, 2010). Rối loạn chuyển hóa carbohydrate, lipid và axit amin xảy ra và góp phần gây ra bệnh béo phì, tiểu đường và NAFLD ở người và ở các sinh vật mô hình [được xem xét trong (Farese và cộng sự, 2012; Lin và Accili, 2011; Newgard, 2012; Samuel và Shulman, 2012; Sun và Lazar, 2013)]. Trong khi các bất thường của tế bào gan trong chuyển hóa lipid tế bào chất thường được quan sát thấy ở NAFLD (Fabbrini và cộng sự, 2010b), vai trò của chuyển hóa ty thể, điều khiển quá trình oxy hóa thải chất béo ít rõ ràng hơn trong cơ chế bệnh sinh NAFLD. Sự bất thường của chuyển hóa ti thể xảy ra và góp phần vào cơ chế bệnh sinh NAFLD / NASH (Hyotylainen và cộng sự, 2016; Serviddio và cộng sự, 2011; Serviddio và cộng sự, 2008; Wei và cộng sự, 2008). Có chung chung (Felig và cộng sự, 1974; Iozzo và cộng sự, 2010; Koliaki và cộng sự, 2015; Satapati và cộng sự, 2015; Satapati và cộng sự, 2012; Sunny và cộng sự, 2011) nhưng không thống nhất ( Koliaki và Roden, 2013; Perry và cộng sự, 2016; Hiệu trưởng và cộng sự, 2010) nhất trí rằng, trước khi có sự phát triển của NASH thực sự, quá trình oxy hóa ty thể ở gan và đặc biệt là quá trình oxy hóa chất béo, làm tăng chứng béo phì, kháng insulin toàn thân. và NAFLD. Có khả năng là khi NAFLD tiến triển, tính không đồng nhất về khả năng oxy hóa, ngay cả giữa các ty thể riêng lẻ, xuất hiện và cuối cùng là chức năng oxy hóa bị suy giảm (Koliaki và cộng sự, 2015; Hiệu trưởng và cộng sự, 2010; Satapati và cộng sự, 2008; Satapati và cộng sự ., 2012).
Ketogenesis thường được sử dụng như một đại diện cho quá trình oxy hóa chất béo ở gan. Sự suy yếu của quá trình tạo xeton xuất hiện khi NAFLD tiến triển trong các mô hình động vật, và có thể ở người. Thông qua các cơ chế chưa được xác định đầy đủ, tăng insulin máu ức chế sự tạo ceton, có thể góp phần gây hạ kali máu so với nhóm chứng gầy (Bergman và cộng sự, 2007; Bickerton và cộng sự, 2008; Satapati và cộng sự, 2012; Soeters và cộng sự, 2009; Sunny và cộng sự. , 2011; Vice và cộng sự, 2005). Tuy nhiên, khả năng dự đoán NAFLD trong cơ thể của nồng độ xeton trong tuần hoàn còn gây tranh cãi (M nnist et al., 2015; Sanyal et al., 2001). Phương pháp quang phổ cộng hưởng từ định lượng mạnh mẽ trên mô hình động vật cho thấy tỷ lệ luân chuyển xeton tăng lên khi kháng insulin vừa phải, nhưng tỷ lệ giảm rõ ràng khi kháng insulin nghiêm trọng hơn (Satapati và cộng sự, 2012; Sunny và cộng sự, 2010). Ở người béo phì có gan nhiễm mỡ, tỷ lệ sinh xeton là bình thường (Bickerton và cộng sự, 2008; Sunny và cộng sự, 2011), và do đó, tỷ lệ tạo xeton giảm đi tương ứng với lượng axit béo tăng lên trong tế bào gan. Do đó, acetyl-CoA có nguồn gốc từ oxy hóa có thể hướng đến quá trình oxy hóa cuối trong chu trình TCA, làm tăng quá trình oxy hóa cuối, tạo gluconeogenesis do phosphoenolpyruvate điều khiển thông qua quá trình anaplerosis / cataplerosis và stress oxy hóa. Acetyl-CoA cũng có thể trải qua quá trình xuất khẩu từ ty thể dưới dạng citrate, một chất nền tiền thân cho quá trình tạo lipogenesis (Hình 4) (Satapati và cộng sự, 2015; Satapati và cộng sự, 2012; Solinas và cộng sự, 2015). Trong khi quá trình tạo ceton trở nên kém đáp ứng với insulin hoặc nhịn ăn khi béo phì kéo dài (Satapati và cộng sự, 2012), các cơ chế cơ bản và hậu quả cuối cùng của điều này vẫn chưa được hiểu đầy đủ. Bằng chứng gần đây chỉ ra rằng mTORC1 ngăn chặn quá trình tạo xeton theo cách có thể là hậu quả của tín hiệu insulin (Kucejova và cộng sự, 2016), phù hợp với các quan sát cho thấy mTORC1 ức chế cảm ứng Hmgcs2 qua PPAR? (Sengupta và cộng sự, 2010) ( cũng xem Quy định của HMGCS2 và SCOT / OXCT1).

Các quan sát sơ bộ từ nhóm của chúng tôi cho thấy hậu quả bất lợi về gan của suy ketogenic (Cotter và cộng sự, 2014). Để kiểm tra giả thuyết rằng suy giảm tạo xeton, ngay cả trong trạng thái dự trữ carbohydrate và do đó, không tạo ketogenic, góp phần vào quá trình chuyển hóa glucose bất thường và gây ra viêm gan nhiễm mỡ, chúng tôi đã tạo ra một mô hình chuột bị suy ketogenic rõ rệt bằng cách sử dụng các oligonucleotide antisense (ASO) nhằm mục tiêu Hmgcs2. Mất HMGCS2 ở chuột trưởng thành được cho ăn ít chất béo tiêu chuẩn gây ra tăng đường huyết nhẹ và tăng rõ rệt việc sản xuất hàng trăm chất chuyển hóa ở gan, một bộ phận trong số đó gợi ý mạnh mẽ đến việc kích hoạt tạo lipid. Chế độ ăn giàu chất béo cho chuột không tạo đủ ketogenesis dẫn đến tổn thương tế bào gan và viêm nhiễm rộng. Những phát hiện này hỗ trợ cho các giả thuyết trung tâm rằng (i) tạo xeton không phải là một con đường tràn thụ động mà là một nút động trong cân bằng nội môi sinh lý tổng hợp và gan, và (ii) tăng sinh xeton thận trọng để giảm NAFLD / NASH và rối loạn chuyển hóa glucose ở gan đáng được thăm dò .
Suy giảm tạo xeton có thể góp phần vào tổn thương gan và thay đổi cân bằng nội môi glucose như thế nào? Việc cân nhắc đầu tiên là liệu thủ phạm có phải là sự thiếu hụt thông lượng xeton hay chính là xeton. Một báo cáo gần đây cho thấy rằng các thể xeton có thể giảm thiểu tổn thương gan do stress oxy hóa khi phản ứng với các axit béo không bão hòa đa n-3 (Pawlak và cộng sự, 2015). Nhớ lại rằng do thiếu biểu hiện SCOT trong tế bào gan, các thể xeton không bị ôxy hóa, nhưng chúng có thể góp phần vào quá trình tạo lipogenesis và phục vụ nhiều vai trò tín hiệu khác nhau độc lập với quá trình ôxy hóa của chúng (cũng xem Số lượng chuyển hóa không ôxy hóa của các thể xeton và? OHB như một trung gian truyền tín hiệu). Cũng có thể các thể xeton có nguồn gốc từ tế bào gan có thể đóng vai trò là tín hiệu và / hoặc chất chuyển hóa cho các loại tế bào lân cận trong vùng gan, bao gồm tế bào hình sao và đại thực bào tế bào Kupffer. Trong khi tài liệu hạn chế có sẵn cho thấy rằng các đại thực bào không thể oxy hóa các thể xeton, điều này chỉ được đo bằng các phương pháp cổ điển và chỉ ở các đại thực bào phúc mạc (Newsholme và cộng sự, 1986; Newsholme và cộng sự, 1987), chỉ ra rằng một tái đánh giá là phù hợp với biểu hiện SCOT phong phú trong các đại thực bào có nguồn gốc từ tủy xương (Youm và cộng sự, 2015).
Thông lượng ketogenic của tế bào gan cũng có thể là chất bảo vệ tế bào. Mặc dù các cơ chế ăn dặm có thể không phụ thuộc vào sự tạo ketogenesis, nhưng chế độ ăn ketogenic ít carbohydrate có liên quan đến việc cải thiện NAFLD (Browning et al., 2011; Foster et al., 2010; Kani et al., 2014; Schugar và Crawford, 2012) . Các quan sát của chúng tôi chỉ ra rằng quá trình tạo xeton tế bào gan có thể phản hồi và điều chỉnh thông lượng chu trình TCA, thông lượng anaplerotic, tạo gluconeogenesis có nguồn gốc từ phosphoenolpyruvate (Cotter và cộng sự, 2014), và thậm chí cả sự luân chuyển glycogen. Suy giảm ketogenic chỉ đạo acetyl-CoA làm tăng thông lượng TCA, trong gan có liên quan đến tăng tổn thương qua trung gian ROS (Satapati và cộng sự, 2015; Satapati và cộng sự, 2012); buộc sự chuyển hướng của carbon vào các loài lipid tổng hợp de novo có thể gây độc tế bào; và ngăn chặn quá trình tái oxy hóa NADH thành NAD + (Cotter và cộng sự, 2014) (Hình 4). Tổng hợp lại, các thí nghiệm trong tương lai được yêu cầu để giải quyết các cơ chế mà qua đó tình trạng suy ketogenic tương đối có thể trở nên tồi tệ, góp phần làm tăng đường huyết, gây viêm gan nhiễm mỡ và liệu các cơ chế này có hoạt động ở NAFLD / NASH ở người hay không. Như bằng chứng dịch tễ học cho thấy sự suy giảm tạo ceton trong quá trình tiến triển của viêm gan nhiễm mỡ (Embade và cộng sự, 2016; Marinou và cộng sự, 2011; M nnist và cộng sự, 2015; Pramfalk và cộng sự, 2015; Safaei và cộng sự, 2016) các liệu pháp làm tăng sự tạo ceton ở gan có thể chứng minh được khả năng ăn mặn (Degirolamo và cộng sự, 2016; Honda và cộng sự, 2016).
Cơ thể Ketone và suy tim (HF)
Với tốc độ trao đổi chất vượt quá 400 kcal / kg / ngày và vòng quay 6-35 kg ATP / ngày, tim là cơ quan có nhu cầu tiêu thụ năng lượng và oxy hóa cao nhất (Ashrafian et al., 2007; Wang et al., 2010b). Phần lớn sự luân chuyển năng lượng của cơ tim nằm trong ty thể, và 70% nguồn cung cấp này bắt nguồn từ FAO. Tim là loài ăn tạp và linh hoạt trong điều kiện bình thường, nhưng tim tái tạo về mặt bệnh lý (ví dụ, do tăng huyết áp hoặc nhồi máu cơ tim) và tim của bệnh nhân tiểu đường trở nên không linh hoạt về mặt trao đổi chất (Balasse và Fery, 1989; BING, 1954; Fukao và cộng sự, 2004 ; Lopaschuk và cộng sự, 2010; Taegtmeyer và cộng sự, 1980; Taegtmeyer và cộng sự, 2002; Young và cộng sự, 2002). Thật vậy, những bất thường được lập trình di truyền của quá trình chuyển hóa nhiên liệu ở tim trong mô hình chuột gây ra bệnh cơ tim (Carley và cộng sự, 2014; Neubauer, 2007). Trong điều kiện sinh lý, tim bình thường sẽ oxy hóa các cơ quan xeton theo tỷ lệ phân phối của chúng, với chi phí là quá trình oxy hóa axit béo và glucose, và cơ tim là cơ quan tiêu thụ xeton cao nhất trên một đơn vị khối lượng (BING, 1954; Crawford et al., 2009; GARLAND et al. ., 1962; Hasselbaink và cộng sự, 2003; Jeffrey và cộng sự, 1995; Pelletier và cộng sự, 2007; Tardif và cộng sự, 2001; Yan và cộng sự, 2009). So với quá trình oxy hóa axit béo, các thể xeton hiệu quả hơn về mặt năng lượng, mang lại nhiều năng lượng hơn cho quá trình tổng hợp ATP trên mỗi phân tử oxy được đầu tư (tỷ lệ P / O) (Kashiwaya và cộng sự, 2010; Sato và cộng sự, 1995; Vectors, 2004) . Quá trình oxy hóa cơ thể xeton cũng tạo ra năng lượng tiềm năng cao hơn FAO, giữ cho ubiquinone bị oxy hóa, làm tăng khoảng cách oxy hóa khử trong chuỗi vận chuyển điện tử và tạo ra nhiều năng lượng hơn để tổng hợp ATP (Sato và cộng sự, 1995; Vectors, 2004). Quá trình oxy hóa các thể xeton cũng có thể hạn chế sản xuất ROS, và do đó gây ra stress oxy hóa (Vectors, 2004).
Nghiên cứu can thiệp và quan sát sơ bộ cho thấy vai trò tiềm năng của các cơ quan ketone trong tim. Trong bối cảnh chấn thương thiếu máu cục bộ / tái tưới máu thực nghiệm, các cơ quan ketone có tác dụng bảo vệ tim mạch tiềm năng (Al-Zaid và cộng sự, 2007; Wang và cộng sự, 2008), có thể là do sự gia tăng sự đa dạng ty thể trong tim hoặc điều hòa sự phosphoryl hóa oxy hóa quan trọng trung gian (Snorek et al., 2012; Zou và cộng sự, 2002). Các nghiên cứu gần đây chỉ ra rằng việc sử dụng cơ thể ketone tăng lên ở tim chuột (Aubert et al., 2016) và con người (Bedi và cộng sự, 2016), hỗ trợ những quan sát trước đây ở người (BING, 1954; Fukao và cộng sự, 2000; Janardhan và cộng sự, 2011; Longo và cộng sự, 2004; Rudolph và Schinz, 1973; Tildon và Cornblath, 1972). Nồng độ ketone lưu hành cơ thể được tăng lên ở bệnh nhân suy tim, tỷ lệ trực tiếp để làm đầy áp lực, quan sát có cơ chế và ý nghĩa vẫn còn chưa biết (Kupari et al., 1995; Lommi et al., 1996; Lommi et al., 1997; Neely et al ., 1972), nhưng những con chuột bị thiếu SCOT có chọn lọc trong các tế bào cơ tim biểu hiện sự tu sửa tâm thất nhanh và các dấu hiệu ROS để đáp ứng với tổn thương quá tải gây áp lực phẫu thuật (Schugar et al., 2014).
Quan sát gần đây hấp dẫn trong điều trị bệnh tiểu đường đã cho thấy một liên kết tiềm năng giữa sự trao đổi chất ketone cơ tim và tu sửa tâm thất bệnh lý (Hình. 5). Sự ức chế vận chuyển đồng vận chuyển natri / glucose dạng ống thận 2 (SGLT2i) làm tăng nồng độ ketone trong cơ thể ở người (Ferrannini và cộng sự, 2016a; Inagaki và cộng sự, 2015) và chuột (Suzuki et al., 2014) qua tăng gan ketogenesis (Ferrannini et al., 2014; Ferrannini et al., 2016a; Katz và Leiter, 2015; Mudaliar và cộng sự, 2015). Đáng chú ý, ít nhất một trong những thuốc này giảm nhập viện HF (ví dụ, như được tiết lộ bởi thử nghiệm EMPA-REG OUTCOME), và cải thiện tử vong tim mạch (Fitchett và cộng sự, 2016; Sonesson và cộng sự, 2016; Wu và cộng sự, 2016a ; Zinman và cộng sự, 2015). Trong khi các cơ chế điều khiển đằng sau kết quả HF có lợi liên quan đến SGLT2i vẫn tích cực tranh luận, lợi ích sống sót có khả năng đa yếu tố, bao gồm cả ketosis, nhưng cũng có tác dụng phụ trên trọng lượng, huyết áp, glucose và mức acid uric, độ cứng động mạch, hệ thần kinh giao cảm, thẩm thấu lợi tiểu / giảm thể tích huyết tương, và tăng hematocrit (Raz và Cahn, 2016; Vallon và Thomson, 2016). Kết hợp với nhau, khái niệm rằng việc tăng ketonemia ở bệnh nhân HF, hoặc những người có nguy cơ cao phát triển HF, vẫn còn gây tranh cãi nhưng đang được điều tra tích cực trong các nghiên cứu tiền lâm sàng và lâm sàng (Ferrannini et al., 2016b; Kolwicz et al., 2016; Lopaschuk và Verma, 2016; Mudaliar và cộng sự, 2016; Taegtmeyer, 2016).

Cơ thể Ketone trong sinh học ung thư
Kết nối giữa các cơ quan ketone và ung thư đang nhanh chóng nổi lên, nhưng các nghiên cứu ở cả hai mô hình động vật và con người đã mang lại kết luận đa dạng. Bởi vì sự trao đổi chất ketone là năng động và đáp ứng trạng thái dinh dưỡng, nó hấp dẫn để theo đuổi các kết nối sinh học với ung thư vì tiềm năng cho các liệu pháp dinh dưỡng có hướng dẫn chính xác. Các tế bào ung thư trải qua quá trình tái lập trình trao đổi chất để duy trì sự phát triển và tăng trưởng tế bào nhanh chóng (DeNicola và Cantley, 2015; Pavlova và Thompson, 2016). Hiệu ứng Warburg cổ điển trong quá trình chuyển hóa tế bào ung thư phát sinh từ vai trò chủ đạo của quá trình lên men glycolysis và axit lactic để truyền năng lượng và bù cho sự phụ thuộc thấp hơn vào quá trình phosphoryl hóa oxy hóa và hô hấp ty thể hạn chế (De Feyter et al., 2016; Grabacka et al., 2016; Kang và cộng sự, 2015; Poff và cộng sự, 2014; Shukla và cộng sự, 2014). Glucose carbon chủ yếu hướng qua glycolysis, con đường phosphate pentose, và lipogenesis, cùng nhau cung cấp trung gian cần thiết cho việc mở rộng sinh khối khối u (Grabacka et al., 2016; Shukla và cộng sự, 2014; Yoshii và cộng sự, 2015). Sự thích nghi của các tế bào ung thư với sự thiếu hụt glucose xảy ra thông qua khả năng khai thác các nguồn nhiên liệu thay thế, bao gồm acetate, glutamine và aspartate (Jaworski và cộng sự, 2016; Sullivan và cộng sự, 2015). Ví dụ, hạn chế truy cập vào pyruvate cho thấy khả năng của các tế bào ung thư để chuyển đổi glutamine thành acetyl-CoA bằng carboxyl hóa, duy trì cả nhu cầu năng lượng và anabolic (Yang et al., 2014). Một sự thích ứng thú vị của các tế bào ung thư là việc sử dụng acetate làm nhiên liệu (Comerford và cộng sự, 2014; Jaworski và cộng sự, 2016; Mashimo và cộng sự, 2014; Wright và Simone, 2016; Yoshii và cộng sự, 2015). Acetate cũng là một chất nền cho lipogenesis, đó là rất quan trọng cho sự phát triển tế bào khối u, và đạt được của ống dẫn lipogenic này có liên quan với sự sống còn bệnh nhân ngắn hơn và gánh nặng khối u lớn hơn (Comerford et al., 2014; Mashimo et al., 2014; Yoshii et al ., 2015).
Các tế bào không phải ung thư dễ dàng chuyển nguồn năng lượng của chúng từ glucose sang thể xeton trong quá trình thiếu hụt glucose. Tính dẻo này có thể thay đổi nhiều hơn giữa các loại tế bào ung thư, nhưng khối u não được cấy ghép in vivo đã bị oxy hóa [2,4-13C2] -? OHB ở mức độ tương tự như mô não xung quanh (De Feyter và cộng sự, 2016). Mô hình Hiệu ứng Warburg ngược hoặc chuyển hóa khối u hai ngăn "đưa ra giả thuyết rằng tế bào ung thư gây ra? Sản xuất OHB trong các nguyên bào sợi lân cận, cung cấp nhu cầu năng lượng của tế bào khối u (Bonuccelli và cộng sự, 2010; Martinez-Outschoorn và cộng sự, 2012) . Ở gan, sự chuyển dịch tế bào gan từ quá trình tạo xeton sang quá trình oxy hóa xeton trong tế bào ung thư biểu mô tế bào gan (u gan) phù hợp với sự hoạt hóa các hoạt động BDH1 và SCOT được quan sát thấy ở hai dòng tế bào u gan (Zhang và cộng sự, 1989). Thật vậy, tế bào u gan biểu hiện OXCT1 và BDH1 và oxy hóa xeton, nhưng chỉ khi huyết thanh bị đói (Huang và cộng sự, 2016). Ngoài ra, quá trình tạo ketogenesis của tế bào khối u cũng đã được đề xuất. Những thay đổi năng động trong biểu hiện gen ketogenic được thể hiện trong quá trình biến đổi ung thư của biểu mô ruột kết, một loại tế bào thường biểu hiện HMGCS2, và một báo cáo gần đây cho thấy HMGCS2 có thể là một dấu hiệu tiên lượng tiên lượng xấu trong ung thư biểu mô tế bào vảy và đại trực tràng (Camarero et al., 2006; Chen và cộng sự, 2016). Cho dù sự liên kết này có yêu cầu hoặc liên quan đến quá trình tạo ketogenesis, hoặc chức năng chiếu sáng mặt trăng của HMGCS2 hay không vẫn còn được xác định. Ngược lại, sản xuất OHB rõ ràng bởi tế bào u ác tính và u nguyên bào thần kinh đệm, được kích thích bởi PPAR? fenofibrate chủ vận, có liên quan đến ngừng tăng trưởng (Grabacka và cộng sự, 2016). Cần có các nghiên cứu sâu hơn để xác định vai trò của biểu hiện HMGCS2 / SCOT, sự tạo xeton và quá trình oxy hóa xeton trong tế bào ung thư.
Ngoài lĩnh vực chuyển hóa nhiên liệu, xeton gần đây còn liên quan đến sinh học tế bào ung thư thông qua cơ chế truyền tín hiệu. Phân tích khối u ác tính BRAF-V600E + chỉ ra cảm ứng HMGCL phụ thuộc OCT1 theo cách phụ thuộc BRAF gây ung thư (Kang và cộng sự, 2015). Việc tăng HMGCL có tương quan với nồng độ AcAc trong tế bào cao hơn, do đó đã tăng cường tương tác BRAFV600E-MEK1, khuếch đại tín hiệu MEK-ERK trong một vòng chuyển tiếp thúc đẩy sự tăng sinh và phát triển của tế bào khối u. Những quan sát này đặt ra câu hỏi hấp dẫn về sự tạo xeton ngoài gan trong tương lai mà sau đó hỗ trợ một cơ chế truyền tín hiệu (cũng xem? OHB như một chất trung gian truyền tín hiệu và Những tranh cãi trong quá trình tạo xeton ngoài gan). Điều quan trọng là phải xem xét các tác động độc lập của AcAc, d-? OHB và l-? OHB đối với chuyển hóa ung thư, và khi xem xét HMGCL, quá trình dị hóa leucine cũng có thể bị lệch lạc.
Tác động của chế độ ăn ketogenic (cũng xem Sử dụng điều trị của chế độ ăn ketogenic và các thể xeton ngoại sinh) trên các mô hình động vật ung thư là rất đa dạng (De Feyter và cộng sự, 2016; Klement và cộng sự, 2016; Meidenbauer và cộng sự, 2015; Poff và cộng sự ., 2014; Seyfried và cộng sự, 2011; Shukla và cộng sự, 2014). Trong khi các mối liên quan dịch tễ học giữa bệnh béo phì, ung thư và chế độ ăn ketogenic đang được tranh luận (Liskiewicz và cộng sự, 2016; Wright và Simone, 2016), một phân tích tổng hợp sử dụng chế độ ăn ketogenic trong mô hình động vật và trong các nghiên cứu trên người cho thấy tác động lớn đến sự sống sót, với lợi ích có liên quan đến mức độ ketosis, thời gian bắt đầu ăn kiêng và vị trí khối u (Klement et al., 2016; Woolf et al., 2016). Điều trị tế bào ung thư tuyến tụy bằng các thể xeton (d-? OHB hoặc AcAc) bị ức chế sự phát triển, tăng sinh và đường phân, và chế độ ăn ketogenic (81% kcal chất béo, 18% protein, 1% carbohydrate) làm giảm trọng lượng khối u in vivo, đường huyết và tăng cơ và trọng lượng cơ thể ở động vật bị ung thư được cấy ghép (Shukla và cộng sự, 2014). Các kết quả tương tự cũng được quan sát bằng cách sử dụng mô hình tế bào u nguyên bào thần kinh đệm di căn ở những con chuột được bổ sung xeton trong chế độ ăn (Poff và cộng sự, 2014). Ngược lại, chế độ ăn ketogenic (91% kcal chất béo, 9% protein) làm tăng nồng độ OHB trong tuần hoàn và giảm đường huyết nhưng không có tác động đến khối lượng khối u hoặc thời gian sống sót ở chuột mang u thần kinh đệm (De Feyter và cộng sự, 2016). Chỉ số xeton glucose đã được đề xuất như một chỉ số lâm sàng giúp cải thiện việc quản lý chuyển hóa của liệu pháp điều trị ung thư não do chế độ ăn ketogenic ở người và chuột (Meidenbauer et al., 2015). Tổng hợp lại, vai trò của chuyển hóa cơ thể xeton và cơ thể xeton trong sinh học ung thư đang trêu ngươi vì chúng đều đưa ra các lựa chọn điều trị có thể kiểm soát được, nhưng các khía cạnh cơ bản vẫn cần được làm sáng tỏ, với những ảnh hưởng rõ ràng xuất hiện từ một ma trận các biến, bao gồm (i) sự khác biệt giữa xeton ngoại sinh cơ thể so với chế độ ăn ketogenic, (ii) loại tế bào ung thư, đa hình gen, cấp độ và giai đoạn; và (iii) thời gian và thời gian tiếp xúc với trạng thái xeton.

Ketogenesis được tạo ra bởi cơ thể ketone thông qua sự phân hủy của các axit béo và axit amin ketogenic. Quá trình sinh hóa này cung cấp năng lượng cho các cơ quan khác nhau, đặc biệt là bộ não, trong các trường hợp ăn chay như là một phản ứng với một không có sẵn của đường huyết. Ketone cơ thể chủ yếu được sản xuất trong ty thể của các tế bào gan. Trong khi các tế bào khác có khả năng thực hiện ketogenesis, chúng không hiệu quả khi làm như tế bào gan. Bởi vì ketogenesis xảy ra trong ti thể, các quá trình của nó được điều chỉnh độc lập. Tiến sĩ Alex Jimenez DC, CCST Insight
Ứng dụng trị liệu của chế độ ăn ketogen và cơ thể Ketone ngoại sinh
Các ứng dụng của chế độ ăn ketogenic và cơ thể xeton làm công cụ điều trị cũng đã phát sinh trong các bối cảnh không ung thư bao gồm béo phì và NAFLD / NASH (Browning và cộng sự, 2011; Foster và cộng sự, 2010; Schugar và Crawford, 2012); suy tim (Huỳnh, 2016; Kolwicz và cộng sự, 2016; Taegtmeyer, 2016); bệnh thần kinh và thoái hóa thần kinh (Martin và cộng sự, 2016; McNally và Hartman, 2012; Rho, 2015; Rogawski và cộng sự, 2016; Yang và Cheng, 2010; Yao và cộng sự, 2011); lỗi bẩm sinh của quá trình trao đổi chất (Scholl-B rgi và cộng sự, 2015); và hiệu suất tập thể dục (Cox và cộng sự, 2016). Hiệu quả của chế độ ăn ketogenic đặc biệt được đánh giá cao trong điều trị động kinh, đặc biệt ở những bệnh nhân kháng thuốc. Hầu hết các nghiên cứu đã đánh giá chế độ ăn ketogenic ở bệnh nhi và cho thấy tần suất co giật giảm tới 50% sau 3 tháng, với hiệu quả được cải thiện trong các hội chứng được chọn (Wu và cộng sự, 2016b). Kinh nghiệm hạn chế hơn ở bệnh nhân động kinh người lớn, nhưng mức giảm tương tự là rõ ràng, với phản ứng tốt hơn ở bệnh nhân động kinh tổng quát có triệu chứng (Nei và cộng sự, 2014). Các cơ chế chống co giật cơ bản vẫn chưa rõ ràng, mặc dù các giả thuyết được đưa ra bao gồm giảm sử dụng glucose / đường phân, vận chuyển glutamate được lập trình lại, tác động gián tiếp đến kênh kali nhạy cảm với ATP hoặc thụ thể adenosine A1, thay đổi biểu hiện đồng dạng kênh natri hoặc ảnh hưởng đến các hormone tuần hoàn bao gồm leptin ( Lambrechts và cộng sự, 2016; Lin và cộng sự, 2017; Lutas và Yellen, 2013). Vẫn chưa rõ liệu tác dụng chống co giật chủ yếu là do cơ thể xeton, hay do hậu quả chuyển hóa theo dòng của chế độ ăn ít carbohydrate. Tuy nhiên, các este xeton (xem bên dưới) dường như làm tăng ngưỡng co giật ở các mô hình động vật bị kích thích co giật (Ciarlone và cộng sự, 2016; D'Agostino và cộng sự, 2013; Viggiano và cộng sự, 2015).
Các chế độ ăn kiêng ít carbohydrate và ketogenic thường được coi là khó chịu và có thể gây táo bón, tăng acid uric máu, hạ calci máu, hạ natri máu, dẫn đến sỏi thận, nhiễm ceton acid, gây tăng đường huyết, tăng cholesterol và nồng độ acid béo tự do (Bisschop et al., 2001) ; Kossoff và Hartman, 2012; Kwiterovich và cộng sự, 2003; Suzuki và cộng sự, 2002). Vì những lý do này, sự tuân thủ lâu dài đặt ra thách thức. Nghiên cứu động vật gặm nhấm thường sử dụng phân phối macronutrient đặc biệt (94% kcal chất béo, 1% kcal carbohydrate, protein 5% kcal, Bio-Serv F3666), trong đó gây ra một ketosis mạnh mẽ. Tuy nhiên, tăng hàm lượng protein, thậm chí đến 10% kcal làm giảm đáng kể ketosis, và hạn chế protein hạn chế 5% kcal làm nhiễu loạn các tác dụng trao đổi chất và sinh lý. Công thức chế độ ăn uống này cũng là cholin cạn kiệt, một biến khác ảnh hưởng đến tính nhạy cảm đối với tổn thương gan, và thậm chí cả ketogenesis (Garbow và cộng sự, 2011; Jornayvaz và cộng sự, 2010; Kennedy và cộng sự, 2007; Pissios và cộng sự, 2013; Schugar et al., 2013). Ảnh hưởng của việc tiêu thụ ketogenic lâu dài ở chuột vẫn chưa được xác định đầy đủ, nhưng những nghiên cứu gần đây trên chuột cho thấy sự sống còn bình thường và không có dấu hiệu gan ở chuột trên chế độ ketogenic trong suốt tuổi thọ của chúng, mặc dù chuyển hóa axit amin, chi tiêu năng lượng và tín hiệu insulin được tái lập trình rõ rệt (Douris et al., 2015).
Cơ chế tăng ketosis thông qua cơ chế thay thế cho chế độ ăn ketogenic bao gồm việc sử dụng tiền chất cơ thể ketone ăn được. Việc quản lý các cơ quan ketone ngoại sinh có thể tạo ra một trạng thái sinh lý duy nhất không gặp phải trong sinh lý bình thường, vì lưu thông glucose và nồng độ insulin là tương đối bình thường, trong khi các tế bào có thể hấp thu và sử dụng glucose. Bản thân các cơ quan xeton có thời gian bán hủy ngắn, và việc uống hoặc truyền muối natri OHB để đạt được trạng thái xeton trị liệu gây ra một lượng natri không đủ tiêu chuẩn. R / S-1,3-butanediol là một chất thẩm tách không độc, dễ bị oxy hóa trong gan để tạo ra d / l-? OHB (Desrochers và cộng sự, 1992). Trong các bối cảnh thí nghiệm khác nhau, liều này đã được sử dụng hàng ngày cho chuột nhắt hoặc chuột cống trong 5 tuần, mang lại nồng độ OHB tuần hoàn lên đến 2 mM trong vòng 3 giờ sau khi dùng, ổn định trong ít nhất 2013 giờ nữa (D ' Agostino và cộng sự, XNUMX). Một phần ức chế ăn uống đã được quan sát thấy trong loài gặm nhấm được đưa ra R / S-1,3-butanediol (Carpenter và Grossman, 1983). Ngoài ra, ba este xeton (KEs), (i) monoester khác biệt về mặt hóa học của R-1,3-butanediol và d-? OHB (R-3-hydroxybutyl R-? OHB); (ii) glyceryl-tris-? OHB; và (iii) R, S-1,3-butanediol acetoacetate diester, cũng đã được nghiên cứu rộng rãi (Brunengraber, 1997; Clarke et al., 2012a; Clarke et al., 2012b; Desrochers et al., 1995a; Desrochers et al. ., 1995b; Kashiwaya và cộng sự, 2010). Một ưu điểm cố hữu của phương pháp trước là 2 mol d- OHB sinh lý được tạo ra trên mỗi mol KE, sau quá trình thủy phân esterase trong ruột hoặc gan. Tính an toàn, dược động học và khả năng dung nạp đã được nghiên cứu rộng rãi nhất ở người uống R-3-hydroxybutyl R-? OHB, với liều lên đến 714 mg / kg, mang lại nồng độ d-? OHB tuần hoàn lên đến 6 mM (Clarke và cộng sự, 2012a; Cox và cộng sự, 2016; Kemper và cộng sự, 2015; Shivva và cộng sự, 2016). Trong động vật gặm nhấm, KE này làm giảm lượng calo và lượng cholesterol tổng số huyết tương, kích thích mô mỡ màu nâu, và cải thiện sức đề kháng insulin (Kashiwaya et al., 2010; Kemper et al., 2015; Veech, 2013). Những phát hiện gần đây chỉ ra rằng trong quá trình tập luyện ở các vận động viên được đào tạo, việc tiêu hóa R-3-hydroxybutyl R-? OHB làm giảm quá trình đường phân ở cơ xương và nồng độ lactate trong huyết tương, tăng quá trình oxy hóa triacylglycerol trong cơ bắp và hàm lượng glycogen trong cơ được bảo toàn, ngay cả khi ăn cùng carbohydrate kích thích tiết insulin Cox và cộng sự, 2016). Cần phát triển thêm các kết quả hấp dẫn này, bởi vì sự cải thiện về hiệu suất tập thể dục bền bỉ chủ yếu được thúc đẩy bởi một phản ứng mạnh mẽ đối với KE trong các đối tượng 2 / 8. Tuy nhiên, những kết quả này hỗ trợ nghiên cứu cổ điển chỉ ra một ưu tiên cho quá trình oxy hóa ketone trên các chất nền khác (GARLAND et al., 1962; Hasselbaink et al., 2003; Stanley và cộng sự, 2003; Valente-Silva và cộng sự, 2015), bao gồm cả trong quá trình luyện tập, và các vận động viên được huấn luyện đó có thể được sử dụng nhiều hơn để sử dụng xeton (Johnson và cộng sự, 1969a; Johnson và Walton, 1972; Winder và cộng sự, 1974; Winder và cộng sự, 1975). Cuối cùng, các cơ chế có thể hỗ trợ hiệu suất tập thể dục được cải thiện sau khi lượng calo bằng nhau (phân bố khác nhau giữa các chất dinh dưỡng) và tỷ lệ tiêu thụ oxy bằng nhau vẫn được xác định.
Quan điểm tương lai
Từng bị kỳ thị phần lớn là một con đường tràn có khả năng tích tụ khí thải độc hại từ quá trình đốt cháy chất béo ở các trạng thái hạn chế carbohydrate (mô hình ketoxic ), các quan sát gần đây ủng hộ quan điểm rằng chuyển hóa cơ thể xeton phục vụ vai trò ăn lương ngay cả trong trạng thái chứa nhiều carbohydrate, mở ra một ketohormetic giả thuyết. Trong khi các phương pháp tiếp cận dinh dưỡng và dược lý dễ dàng để điều khiển chuyển hóa xeton khiến nó trở thành mục tiêu điều trị hấp dẫn, các thí nghiệm được đặt ra mạnh mẽ nhưng thận trọng vẫn được duy trì trong cả phòng thí nghiệm nghiên cứu cơ bản và dịch thuật. Các nhu cầu chưa được đáp ứng đã xuất hiện trong các lĩnh vực xác định vai trò của việc tận dụng chuyển hóa xeton trong bệnh suy tim, béo phì, NAFLD / NASH, bệnh tiểu đường loại 2 và ung thư. Phạm vi và tác động của các vai trò tín hiệu 'không chuẩn' của các cơ quan xeton, bao gồm điều chỉnh các PTM có khả năng truyền ngược và chuyển tiếp vào các con đường chuyển hóa và tín hiệu, đòi hỏi phải khám phá sâu hơn. Cuối cùng, quá trình tạo xeton ngoài gan có thể mở ra các cơ chế truyền tín hiệu nội tiết và nội tiết hấp dẫn và các cơ hội để tác động đến đồng chuyển hóa trong hệ thần kinh và khối u để đạt được mục đích điều trị.
Lời cảm ơn
Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/
Chú thích
Kết luận, các cơ quan xeton được tạo ra bởi gan để được sử dụng làm nguồn năng lượng khi cơ thể con người không có đủ glucose. Quá trình tạo xeton xảy ra khi có mức đường huyết thấp, đặc biệt là sau khi các kho dự trữ carbohydrate tế bào khác đã cạn kiệt. Mục đích của bài viết trên là thảo luận về vai trò đa chiều của các thể xeton trong chuyển hóa nhiên liệu, truyền tín hiệu và điều trị. Phạm vi thông tin của chúng tôi được giới hạn trong các vấn đề sức khỏe cột sống và thần kinh cột sống. Để thảo luận về chủ đề này, vui lòng hỏi Tiến sĩ Jimenez hoặc liên hệ với chúng tôi theo địa chỉ 915-850-0900 .
Quản lý bởi Tiến sĩ Alex Jimenez
Được tham chiếu từ: Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/

Thảo luận về chủ đề bổ sung: Đau lưng cấp tính
đau lưng là một trong những nguyên nhân phổ biến nhất của tình trạng khuyết tật và bỏ lỡ ngày làm việc trên toàn thế giới. Đau lưng thuộc về lý do phổ biến thứ hai để đến phòng khám bác sĩ, chỉ nhiều hơn do nhiễm trùng đường hô hấp trên. Khoảng 80 phần trăm dân số sẽ bị đau lưng ít nhất một lần trong suốt cuộc đời của họ. Cột sống là một cấu trúc phức tạp được tạo thành từ xương, khớp, dây chằng và cơ cùng với các mô mềm khác. Thương tích và / hoặc tình trạng trầm trọng hơn, chẳng hạn nhưđĩa đệm thoát vị, cuối cùng có thể dẫn đến các triệu chứng đau lưng. Chấn thương thể thao hoặc chấn thương do tai nạn ô tô thường là nguyên nhân gây đau lưng thường xuyên nhất, tuy nhiên, đôi khi những động tác đơn giản nhất cũng có thể gây đau đớn. May mắn thay, các lựa chọn điều trị thay thế, chẳng hạn như chăm sóc thần kinh cột sống, có thể giúp giảm đau lưng thông qua việc điều chỉnh cột sống và thao tác bằng tay, cuối cùng là cải thiện việc giảm đau.

EXTRA EXTRA | CHỦ ĐỀ QUAN TRỌNG: Khuyến nghị El Paso, TX Chiropractor
***